

 产业资讯
产业资讯
 精准药物
精准药物  2023-09-08
2023-09-08
 4054
4054
据公开资料显示,2023年7-8月,NMPA批准了包含靶向HER2的ADC药物-德曲妥珠单抗、EGFR抑制剂-舒沃替尼等在内的3项肿瘤药物适应症,FDA也批准了多达6项肿瘤药物适应症。详细7-8月获批上市药物及药物新获批适应症见下文。
Part.1
新药速递(概括版)

Part.2
新药速递(详细版)
1. 赛帕利单抗 /
商品名:誉妥
通用名:赛帕利单抗、Zimberelimab
适应症:PD-L1表达阳性的宫颈癌患者
临床试验:NCT03972722
原研公司:誉衡生物
获批日期:2023.07.04
获批机构:NMPA
7月4日,根据NMPA官网最新公示,誉衡生物PD-1抑制剂赛帕利单抗的新适应症上市申请已获得批准。这是一款全人源抗PD-1单抗,已于2021年8月在中国获批用于复发或难治性经典霍奇金淋巴瘤治疗。公开资料显示,此次为赛帕利单抗获批的第二项适应症,用于接受过一线或以上含铂标准化疗后进展的复发或转移、PD-L1表达阳性(CPS≥1)的宫颈癌患者的治疗。
临床数据:本次适应症的获批是基于一项II期关键性注册临床研究结果(NCT03972722)。该II期临床研究数据已由吴小华教授团队在多个国际知名学术会议上发表,包括2022年第37届癌症免疫治疗学会(SITC)年会电子壁报,2022年欧洲肿瘤内科学会免疫肿瘤学(ESMO IO)大会电子壁报,及2022年欧洲肿瘤内科学会亚洲年会(ESMO ASIA)的口头报告。
根据2023年3月誉衡生物新闻稿中披露的研究结果,截至2022年4月29日,在全分析集中,赛帕利单抗单药治疗的客观缓解率(ORR)达到27.8%,其中5例患者获得完全缓解(CR),20例获得部分缓解(PR);中位总生存期(OS)达到16.8个月,中位缓解持续时间(DoR)尚未达到。此外,赛帕利单抗展现了长期疗效和较高的客观缓解率,且安全性良好。

赛帕利单抗公开研究信息
赛帕利单抗是全球第一个使用国际先进的转基因大鼠平台(OmniRat®)自主研发的全人源抗PD-1单克隆抗体。2021年8月25日,本品的首个针对复发或难治性经典型霍奇金淋巴瘤的适应症获批,并于2020-2022年连续3年入选《CSCO淋巴瘤诊疗指南》,并获得专家的I级推荐;2021年3月,赛帕利单抗因针对宫颈癌的疗效数据突出,而纳入药品审评中心突破性治疗品种名单。这一产品通过阻断PD-1信号通路来激发人体自身的免疫系统,清除患者体内的癌细胞,从而实现抗肿瘤作用。公开资料显示,赛帕利单抗与PD-1的结合位点位于PD-1的C strand、FG loop和G strand,与PD-L1/PD-1的结合区域十分接近,有望全方位阻断PD-1与PD-L1的结合。此外,它还经过特殊修饰从而克服IgG4不稳定性引起的疗效和毒性的不可预测性。
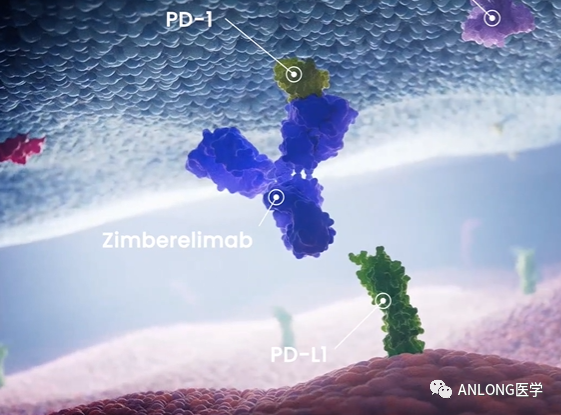
赛帕利单抗作用机制
2. 德曲妥珠单抗 /
商品名:优赫得、Enhertu
通用名:德曲妥珠单抗、Trastuzumab Deruxtecan
适应症:HER2低表达乳腺癌患者
临床试验:DESTINY-Breast04
原研公司:阿斯利康&第一三共
获批日期:2023.07.12
获批机构:NMPA
7月12日,阿斯利康(AstraZeneca)和第一三共(Daiichi Sankyo)宣布,双方联合开发和商业化的靶向HER2的ADC药物-德曲妥珠单抗获得NMPA正式批准,本品单药适用于治疗既往在转移性疾病阶段接受过至少一种系统治疗的,或在辅助化疗期间或完成辅助化疗之后6个月内复发的,不可切除或转移性HER2低表达成人乳腺癌患者。
这是继2023年2月首次在华获批单药治疗既往接受过一种或一种以上抗HER2药物治疗的不可切除或转移性HER2阳性成人乳腺癌患者后,德曲妥珠单抗在中国获批的又一重磅适应症。该获批疗法为国内首个有助于HER2低表达转移性乳腺癌患者延缓疾病进展和延长生存期的创新靶向疗法,有望重塑HER2乳腺癌分型和治疗。

德曲妥珠单抗在国内外获批癌种
临床数据:此次获批是基于DESTINY-Breast04 III期临床试验的积极结果,这是一项全球性、随机、开放标签、注册试验,该研究是首个针对HER2低表达转移性乳腺癌患者的临床试验,纳入HER2低表达、不可切除、和/或转移性乳腺癌患者,这些患者既往接受过1-2线化疗,按2:1随机分组,分为德曲妥珠单抗组和化疗组。结果显示,HR阳性人群中,德曲妥珠单抗组和化疗组的无进展生存期(PFS)分别为10.1个月和5.4个月;总人群中,德曲妥珠单抗组和化疗组的PFS分别为9.9个月和5.1个月。OS方面,HR阳性人群中,德曲妥珠单抗组和化疗组的OS分别为23.9个月和17.5个月;总人群中,德曲妥珠单抗组和化疗组的OS分别为23.4个月和16.8个月。
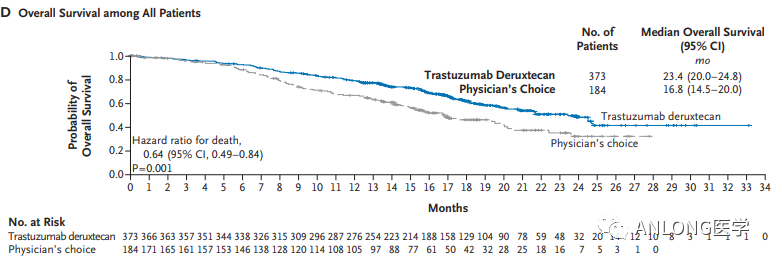
DESTINY-Breast04临床试验OS数据
乳腺癌是威胁全球女性健康的第一大恶性肿瘤,2020年中国确诊乳腺癌病例已接近42万,其中,HER2低表达的乳腺癌患者占全部类型的45%-55%,但由于缺乏针对性的靶向治疗选择,HER2低表达的患者常被确诊为HER2阴性乳腺癌,而在传统治疗手段下,患者的生存时间和生存质量仍面临诸多挑战。此次德曲妥珠单抗在中国获批,告别了HER2阳性和阴性二分法时代,为HER2低表达乳腺癌患者提供了全新的治疗选择。目前ASCO和NCCN指南均将德曲妥珠单抗作为HER2低表达晚期乳腺癌患者的优选治疗推荐,同时2023 ESMO指南进一步明确了德曲妥珠单抗的临床应用场景。
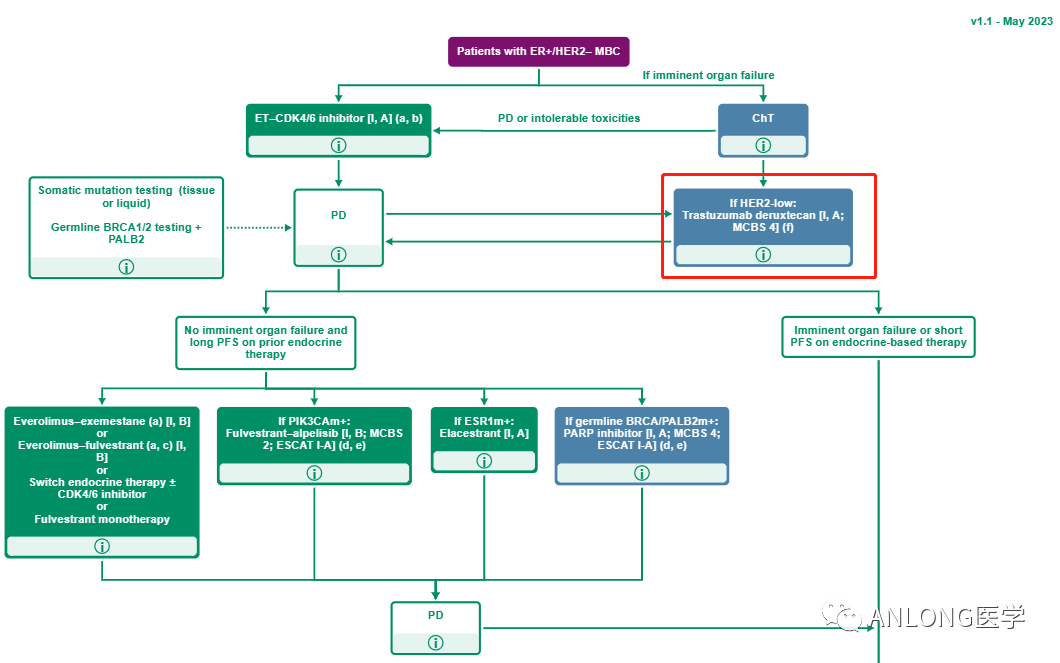
2023. v1.1 ESMO乳腺癌指南
3. Quizartinib /
商品名:Vanflyta
通用名:Quizartinib
适应症:FLT3-ITD阳性的AML患者
临床试验:QuANTUM-First
原研公司:第一三共
获批日期:2023.07.20
获批机构:FDA
7月20日,第一三共宣布,FDA已批准Quizartinib治疗新诊断FLT3-ITD阳性的AML成人患者的上市申请,该药物可联合用于标准的阿糖胞苷+蒽环类药物的诱导缓解治疗和阿糖胞苷巩固治疗,以及单药用于巩固后的持续治疗。
临床数据:此项批准是基于2022 EHA大会上公布的QuANTUM-First研究数据。QuANTUM-First是一项随机、双盲、安慰剂对照的III期研究,旨在评估Quizartinib在新诊断FLT3-ITD阳性AML成人患者中的OS改善情况。这些患者按1:1随机分配,接受Quizartinib或安慰剂联合标准的阿糖胞苷+蒽环类药物的诱导缓解治疗和阿糖胞苷巩固治疗,以及巩固后的单药治疗。研究的主要终点是OS,次要终点包括无事件生存期(EFS)、诱导后CR和复合CR等。
结果显示,在中位随访39.2个月时,Quizartinib组的中位OS为31.9个月,而安慰剂组为15.1个月,这表明Quizartinib联合化疗在携带FLT3-ITD突变的AML成人患者中实现了显著的OS获益。
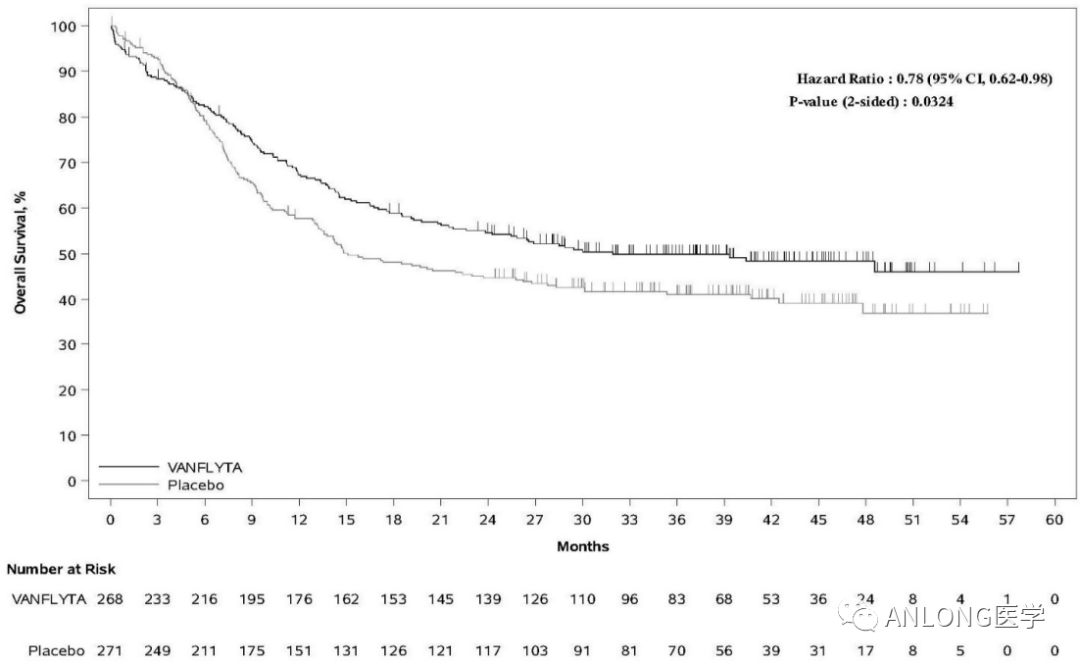
QuANTUM-First临床试验OS数据
4. Dostarlimab-gxly /
商品名:Jemperli
通用名:Dostarlimab-gxly
适应症:dMMR/MSI-H的子宫内膜癌患者
临床试验:RUBY
原研公司:葛兰素史克(GSK)
获批日期:2023.07.31
获批机构:FDA
7月31日,GSK宣布FDA已批准Dostarlimab-gxly联合卡铂和紫杉醇后,接续Dostarlimab-gxly单药治疗dMMR/MSI-H的原发性晚期或复发性子宫内膜癌成年患者。Dostarlimab-gxly是首款获FDA批准用于dMMR/MSI-H子宫内膜癌一线治疗的PD-1抑制剂。此前,该药已获批用于此类子宫内膜癌患者的二线治疗。
临床数据:本次获批主要是基于III期RUBY研究(NCT03981796)期中分析结果。结果显示,在超过25个月中位随访时间内,该试验达到了研究者评估的PFS主要终点,接受Dostarlimab-gxly组合疗法的dMMR/MSI-H患者,中位PFS为30.3个月,对照组的中位PFS为7.7个月。结果表明,接受Dostarlimab-gxly联合卡铂和紫杉醇治疗的dMMR/MSI-H患者的PFS在统计学意义和临床意义上延长。此外,患者经治后,疾病进展或死亡风险降低了71%。意向性治疗(ITT)人群的OS是该研究的另一共同主要终点,目前正在评估中。
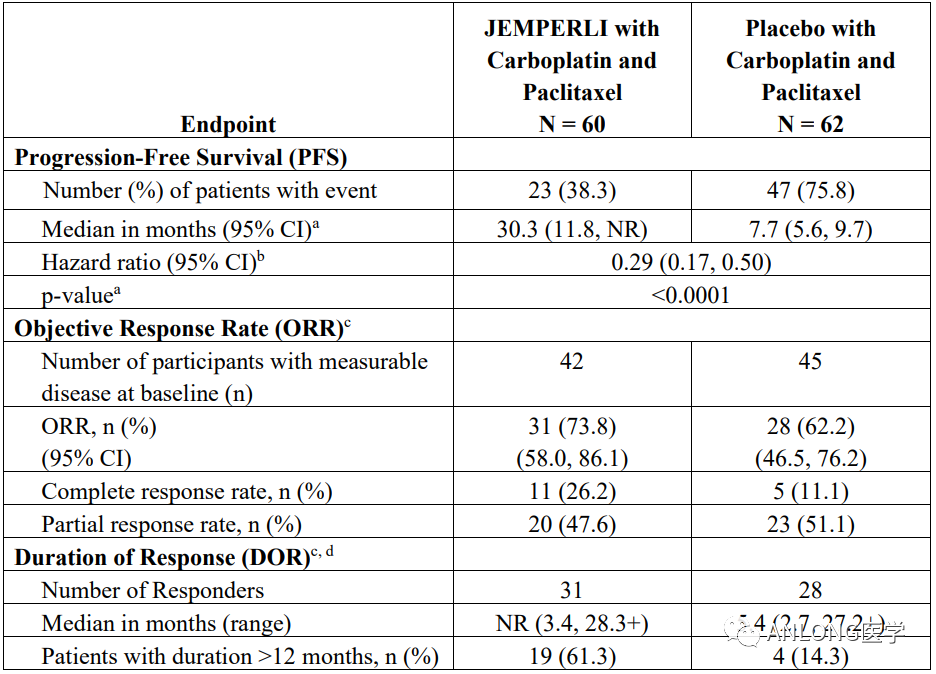
RUBY研究临床数据
5. 普拉替尼 /
商品名:普吉华、Gavreto
通用名:普拉替尼、Pralsetinib
适应症:RET阳性NSCLC
临床试验:ARROW
原研公司:Blueprint Medicines
获批日期:2023.08.09
获批机构:FDA
普拉替尼于2020年9月基于ARROW(NCT03037385)临床试验结果获得美国FDA加速批准用于治疗RET融合阳性非小细胞肺癌成年患者。本次是FDA基于新增的123名患者以及25个月额外随访的数据将“加速批准”转为“常规批准”。
临床数据:ARROW试验共纳237例局部晚期或转移性RET融合阳性NSCLC患者,所有患者接受普拉替尼治疗,直到疾病进展或出现不可接受的毒性。在107例初治患者中,ORR为78%,中位DoR为13.4个月。在既往接受铂类化疗的130例患者中,ORR为63%,中位DoR为38.8个月。
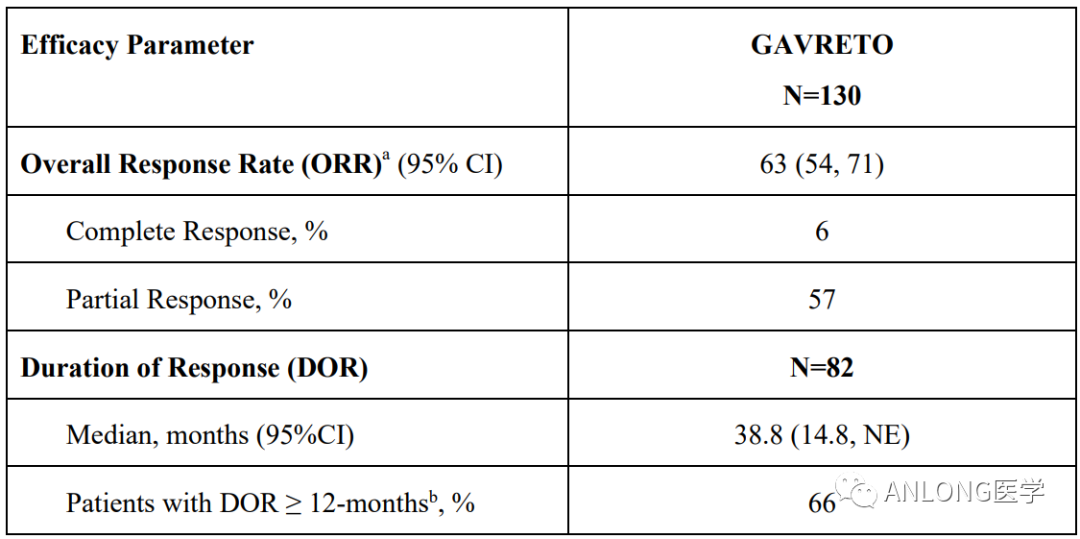
ARROW试验中既往接受铂类化疗的患者临床数据
此外,该研究纳入了来自10个中国研究中心的68例晚期RET融合阳性NSCLC患者,结果显示,不管既往是否接受过治疗,普拉替尼在中国RET融合阳性晚期NSCLC患者中均取得了持久的临床获益,并且初治患者的缓解率更高。临床结果显示,在既往接受过铂类化疗的患者中,确认的ORR为66.7%,包括1例CR和21例PR,疾病控制率(DCR)为93.9%。在未接受过系统性治疗的患者中,确认的ORR为83.3%,包括2例CR和23例PR,DCR为86.7%。
6. Talquetamab-tgvs /
商品名:Talvey
通用名:Talquetamab-tgvs
适应症:R/R MM患者
临床试验:MonumenTAL-1
原研公司:杨森
获批日期:2023.08.10
获批机构:FDA
8月10日,强生宣布,FDA已加速批准其GPRC5D/CD3双抗TALVEY(Talquetamab-tgvs),用于治疗R/R MM成人患者,这些患者此前至少接受过四种疗法,包括蛋白酶体抑制剂、免疫调节剂和CD38抗体。MM是一种无法治愈的血癌,可影响一种称为浆细胞的白细胞,浆细胞存在于骨髓中。在MM中,这些浆细胞发生变化,迅速扩散并以肿瘤取代骨髓中的正常细胞。
临床数据:本次获批是基于MonumenTAL-1的I/II期临床研究,该试验纳入了既往接受过至少4线治疗且既往未接受过T细胞重定向治疗的患者(n=187),结果显示了有意义的ORR。在每两周一次0.8 mg/kg皮下(SC)注射剂量下,73.6%的患者达到ORR。在每周一次0.4 mg/kg SC剂量下,73.0%的患者达到ORR。
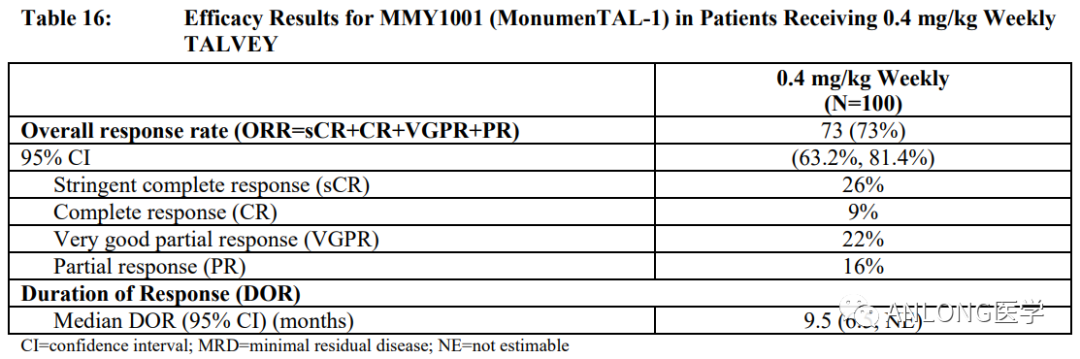
MonumenTAL-1试验临床数据(每周一次0.4 mg/kg SC剂量)
MonumenTAL-1研究还纳入了32例既往暴露于双特异性抗体或CAR-T细胞治疗(94%为B细胞成熟抗原(BCMA)靶向治疗)且既往接受过至少4线治疗(包括蛋白酶体抑制剂、免疫调节剂和抗CD38单克隆抗体)的患者,每周一次接受Talquetamab-tgvs 0.4 mg/kg SC给药。中位随访时间为10.4个月,根据独立审评委员会的评估,72%的患者达到ORR,估计59%的缓解者维持缓解至少9个月。
Talquetamab-tgvs是一种“first-in-class”的双特异性T细胞结合抗体,可结合T细胞表面表达的CD3受体和GPRC5D受体,这是一种新型MM靶标,在MM细胞和非恶性浆细胞以及一些健康组织如皮肤和舌上皮细胞表面高度表达。2021年5月和2021年8月,Talquetamab-tgvs分别被美国FDA和欧盟委员会授予治疗MM的孤儿药资格。Talquetamab-tgvs也于2022年6月获得美国美国FDA突破性疗法认定,用于既往接受过至少4线治疗的R/R MM成人患者的治疗。
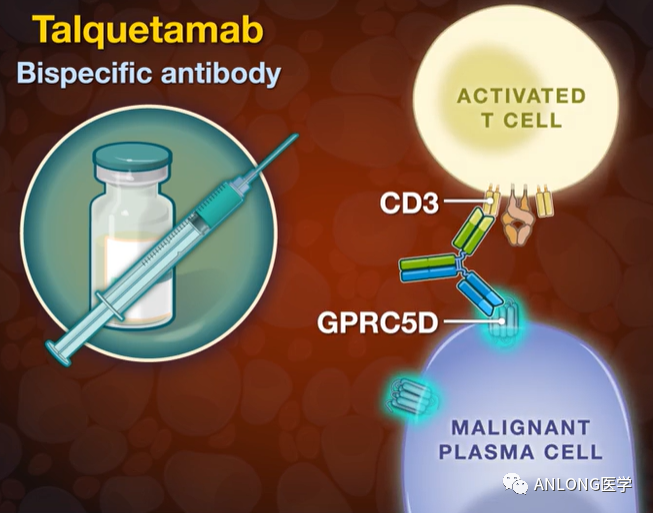
Talquetamab-tgvs作用机制
7. 尼拉帕利+醋酸阿比特龙 /
商品名:Akeega
通用名:尼拉帕利+醋酸阿比特龙、Niraparib + Abiraterone acetate
适应症:mCRPC患者
临床试验:MAGNITUDE
原研公司:强生
获批日期:2023.08.11
获批机构:FDA
8月11日,FDA批准尼拉帕利与醋酸阿比特龙上市,联合泼尼松用于治疗有害或疑似有害BRCA突变的mCRPC患者。这是FDA批准的首个也是唯一一个用于该适应症的双作用片剂。
临床数据:本次批准是基于III期MAGNITUDE研究的结果。MAGNITUDE是一项随机、双盲、安慰剂对照的III期临床试验,旨在评估尼拉帕利联合醋酸阿比特龙和泼尼松(AAP)对比安慰剂联合AAP一线治疗有或无同源重组修复(HRR)基因突变的mCRPC患者的疗效和安全性,共纳入423例患者(其中225例患者携带BRCA突变),主要终点为影像学无进展生存期(rPFS)。
结果显示,在BRCA突变患者中,尼拉帕利+AAP较安慰剂+AAP显著改善了患者的rPFS,两组患者的中位rPFS分别为16.6个月和10.9个月。此外,在BRCA突变患者中,尼拉帕利+AAP组患者的中位OS也更加具有改善趋势,两组患者的中位OS分别为30.4个月和28.6个月。
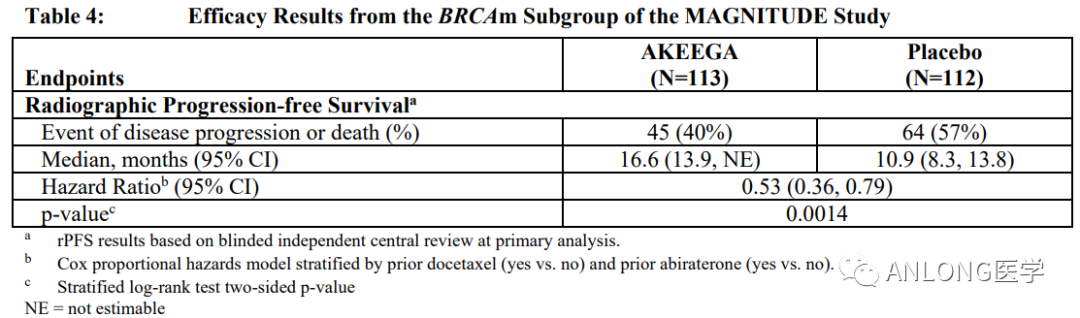
MAGNITUDE试验临床数据
8. Elranatamab-bcmm /
商品名:Elrexfio
通用名:Elranatamab-bcmm
适应症:R/R MM患者
临床试验:MagnetisMM-3
原研公司:辉瑞
获批日期:2023.08.14
获批机构:FDA
8月14日,辉瑞宣布其研发的CD3/BCMA双抗Elranatamab-bcmm获FDA加速批准上市,用于治疗R/R MM,这些患者既往至少接受过四种治疗,包括蛋白酶体抑制剂、免疫调节剂和抗CD38单克隆抗体。这也是继强生Tecvayli(Teclistamab-cqyv)后,第二款获FDA批准上市的CD3/BCMA双抗。
临床数据:Elranatamab-bcmm是一种人源化的双特异性抗体,能同时靶向表达BCMA的MM细胞和表达CD3的T细胞。本次获批是基于MagnetisMM-3(NCT04649359)研究队列A的积极数据。该研究是一项开放标签、多中心、单臂的II期研究,旨在评估Elranatamab-bcmm单药治疗R/R MM的安全性和有效性。其中队列A入组了123例至少对1种蛋白酶体抑制剂(PI)、1种免疫调节药物(IMiD)和1种抗CD38单抗耐药的R/R MM患者。
在2023年欧洲血液学协会会议上提交的队列A(n=123)的长期疗效数据显示,中位随访10.4个月时,接受Elranatamab-bcmm作为首次BCMA靶向治疗的患者ORR为61%,超过56.1%的患者达到了非常好的部分缓解率(VGPR)。在为期14.7个月的中位随访中,中位DoR、PFS及OS尚未达到。对于出现缓解的患者,在15个月时维持缓解的概率为72%。此外,研究中Elranatamab-bcmm具有可控的安全性。
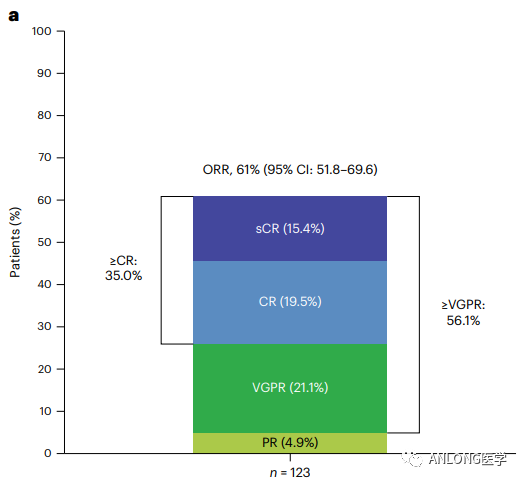
MagnetisMM-3试验临床数据
9. 舒沃替尼 /
商品名:舒沃哲
通用名:舒沃替尼、Sunvozertinib
适应症:EGFR exon20ins的NSCLC患者
临床试验:WU-KONG6
原研公司:迪哲医药
获批日期:2023.08.22
获批机构:NMPA
2023年8月23日,上海迪哲医药宣布,公司首款自主研发的新型肺癌靶向药舒沃哲(通用名:舒沃替尼片)于8月22日正式获得NMPA批准,用于既往经含铂化疗治疗后出现疾病进展,或不耐受含铂化疗,并且经检测确认存在EGFR exon20ins的局部晚期或转移性NSCLC患者。舒沃替尼成为首款针对EGFR exon20ins突变型晚期NSCLC的国创新药。
临床数据:舒沃替尼是一款针对多种EGFR突变亚型的高选择性EGFR靶向药物,此次获批主要基于中国注册临床试验(悟空6,WU-KONG6),该研究是一项针对含铂化疗进展或不耐受的EGFR exon20ins晚期NSCLC患者的单臂、多中心II期注册研究,主要终点为独立影像评估委员会(IRC)评估的ORR,研究结果在2023年ASCO年会上以口头报告形式公布。
在接受舒沃替尼治疗的97例疗效分析人群中,经IRC确认的ORR达60.8%,超过90%的患者经舒沃替尼单药治疗后靶病灶缩小,DCR达87.6%。至数据截止日期,64.4%(38/59)的患者仍处于持续缓解中,DoR仍未达到,最长DoR已超过11.2个月。安全性与传统EGFR-TKI相似,整体耐受性好,临床可管理可恢复。舒沃替尼凭借高效低毒有望成为EGFR exon20ins突变晚期NSCLC患者更优治疗选择。
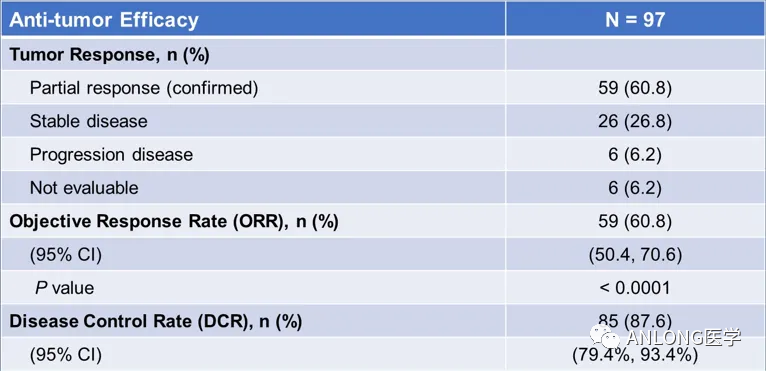
IRC评估的舒沃替尼的抗肿瘤疗效
 产业资讯
药融圈 2025-07-07
111
产业资讯
药融圈 2025-07-07
111
 产业资讯
医药魔方Pro 2025-07-07
115
产业资讯
医药魔方Pro 2025-07-07
115
 产业资讯
药研网 2025-07-07
116
产业资讯
药研网 2025-07-07
116
 微信公众号
微信公众号 热门资讯
热门资讯 热点标签
热点标签