

 产业资讯
产业资讯
 研发客
研发客  2023-11-02
2023-11-02
 2455
2455
10月30日,君实生物自主研发的PD-1单抗药物特瑞普利单抗的生物制品许可申请(BLA)获得FDA批准,再次引发业内对出海美国的的讨论热点。恰好,研发客和太美医疗联合举办了“中国如何出海做临床”的研讨会,与会嘉宾围绕为何出海、不同时间点需要注意什么等方面进行了讨论。我们将会议讨论的精华整理成文,供读者参考。
临床开发 Vs. 临床试验
拓创生物创始人Mann Fung博士在美国FDA、强生和礼来有多年临床开发经验,也熟悉美国文化。他强调临床开发(Clinical Development)和临床试验(Clinical Trails)的定义有所不同。

Mann Fung博士
“临床开发是一家公司整体开发战略,关注药品全生命周期,而临床试验只是某一具体项目,关注短期和单个试验目标、临床终点,主要为项目管理。”他强调,中国公司首先要有战略规划,其次要对操作细节进行把握。
对于中国公司而言,美国为何仍是临床试验最佳的胜地?冯博士展示了美国临床试验的全景图(见下图)。
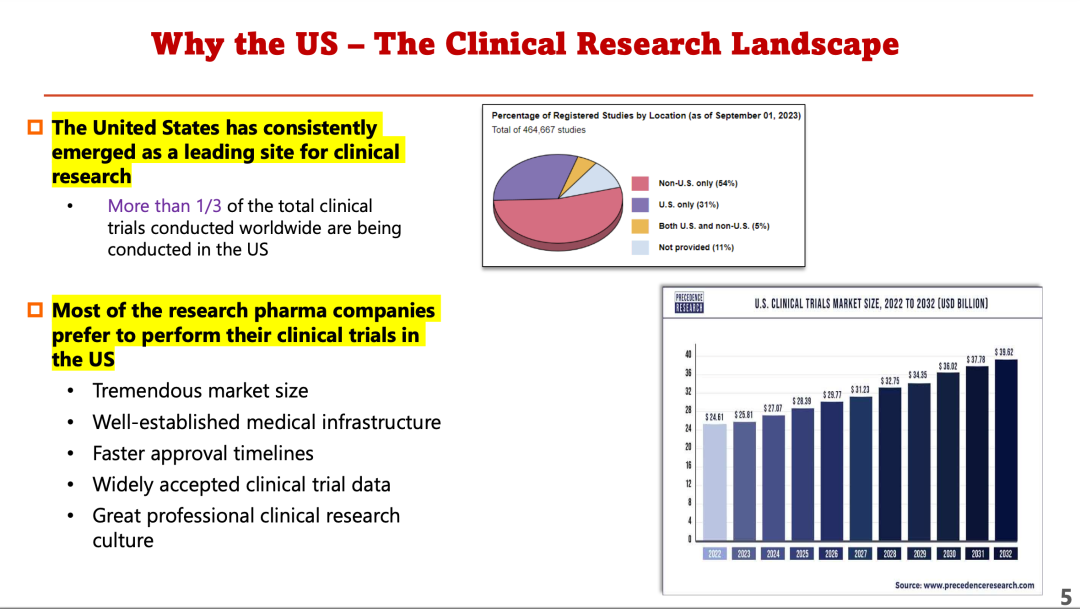
要成功在美国开展临床试验,必须注意几个因素:
首先,经费很重要。如果没有资金,试验难以为继。他举例说,在美国,成功完成从临床试验到获批上市的药物研发全生命流程平均需要26亿美元。22%的试验都在3期因没有足够费用而达不到在预定疗效上证明具有统计学显著意义所需的患者人数。
其次是开展合作,如授权转让、联合开发等。
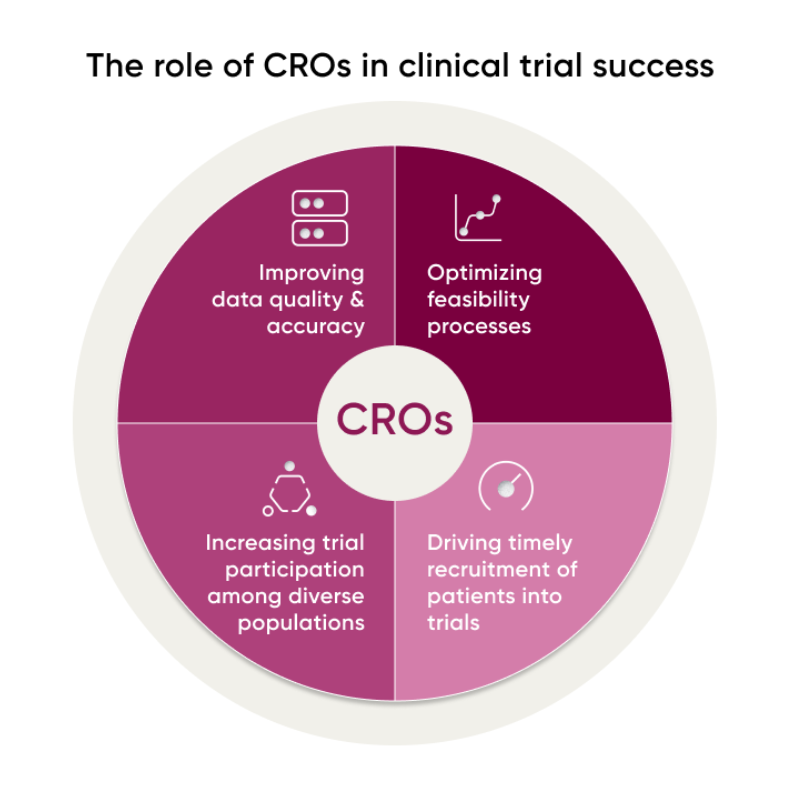
第三是找到一个专业、可信赖的CRO(其作用如下图)。
第四是患者招募。美国大约33%的试验因未能达到招募目标而延时。25%的肿瘤试验未能招募足够患者;18%的试验参与人数不到目标受试者的一半。由于现在研究设计中,入组标准越来越高,导致难以找到合适的受试者,也令试验迟迟不能开始。
第五是FDA强调患者人群多样性。去年2月10日,FDA的ODAC会议拒绝信迪利单抗3期试验ORIENT-11仅基于中国患者数据在美国上市,认为该试验不足以反映美国医疗实践。索凡替尼也因同样理由被FDA拒批。不过,中国企业已吃一堑长一智。
第六是CM&C和稽查核查问题。FDA最近因质量问题拒批大小药厂的案例不绝于耳。能否恒久保持药品质量是FDA关注的重点。
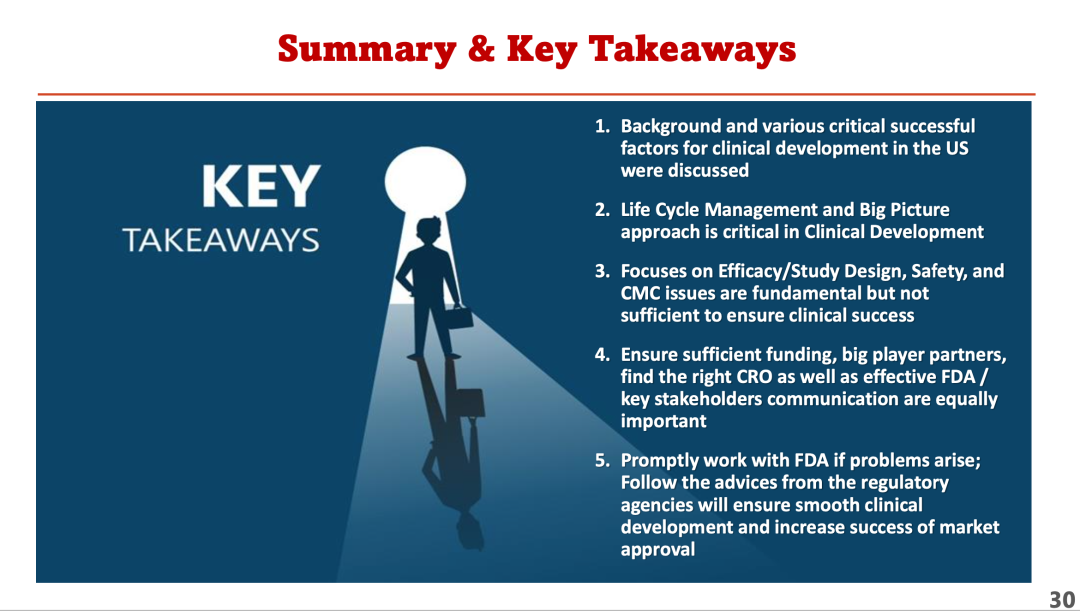
最后是企业与FDA、研究医院、CRO等的协调能力,这也是中国企业最大的挑战之一。冯博士列举了阿斯利康5R等经典模式,最后得出5个结论(见下图)。
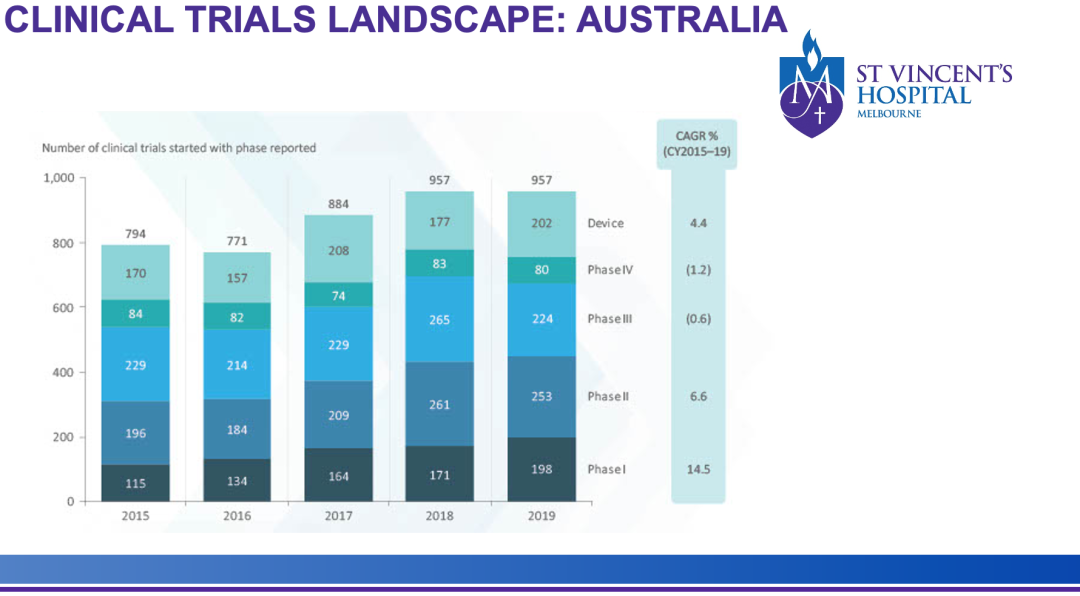
澳洲数据如何用于申报FDA
近年来,澳大利亚(以下简称“澳洲”)已成为有计划到美国发展的中国公司开展1期临床的首选地。
墨尔本圣文森特(St Vincent’s)医院研究副主任、墨尔本医学院副教授Tam C. Nguyen博士特别讲解了澳洲1期临床数据用于支持美国启动 2 期研究和被FDA接受的注意事项(具体见如下归纳)。

Tam C. Nguyen博士
他认为,仔细规划符合监管要求及与FDA沟通,对确保FDA成功接受澳洲数据至关重要。
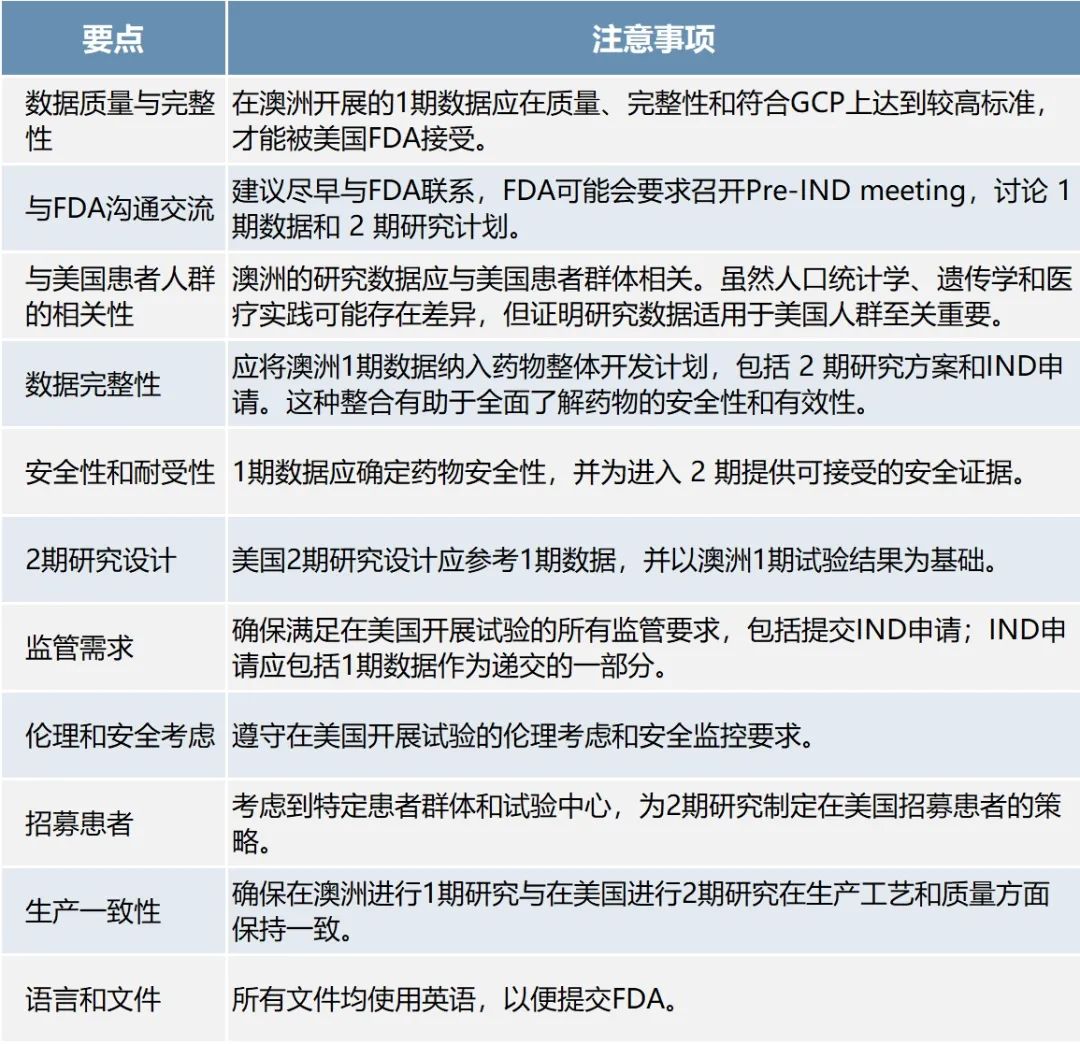
整理自Tam C. Nguyen博士的PPT
他举了一个例子,一家小型生物制药公司PharmaBio开发了一种治疗罕见的自身免疫疾病创新药TheraX。由于临床前研究结果良好和澳洲的种种优势,该公司希望在该国启动1期试验加快开发。1期试验对 TheraX 在健康志愿者中的安全性、给药方案、药动学和药效学提供了重要的数据,并为2期临床开发提供了决策依据。PharmaBio与FDA开展了早期、透明和充分的沟通,分享了澳洲的1期数据,并展示其与 FDA 标准的一致性。最后,IND 申请的成功凸显了在澳洲开展 1 期试验的优势和重要性。
从IND到NDA的美国整体计划
前FDA资深临床审评员、思路迪医药首席医学官肖申博士也赞同申请人要有整体试验设计方案,清晰地知道在美国开展试验的目的和针对人群,试验方案的要点包括受试者入选标准、给药方案、安全监控、终止标准等。

肖申博士
在患者选择上,须满足人群多样性,并符合美国的医疗实践。在试验设计方法学上,FDA近年来结合工业界出现的新变化,出台了包括富集试验、适应性设计等,申请人可以灵活运用,但要积极跟FDA提前沟通。“FDA非常重视企业的计划性和前瞻性。”肖申说。

在终点选择上,为了降低研发难度和节省时间和成本,FDA也会允许企业选择替代终点。尤其是肿瘤药研发,近些年,总生存期(OS)已被客观缓解率(ORR)和无进展生存期(PFS)替代,成为临床试验的终点。然而,肖申说,有很多证据证明,替代终点与OS之间相关性并不紧密,也不确定。
在鼓励创新方面,除了业内熟悉的突破性疗法、快速审批等政策,FDA还在肿瘤药方面推出了4项新举措,包括Project Front Runner、Project Orbis、Project Optimus、Project Equity(具体内容请留意研发客对肖申的专访)。
申请人往往希望快速推进临床试验,但需要注意FDA会因为非临床和临床的种种不足,发出Clinical Hold的指令。而不同的Clinical Hold会带来不同后果,对上市公司而言,一定要谨慎,尽可能不要被FDA“Hold”住而影响了公司股价。

最后,在NDA递交和审评环节,有几个细节值得注意。Pre-NDA meeting是决定产品能否获批的最后一道关卡,企业要将最全面、完整的数据在60天内递交给FDA。到了第74天,FDA会给申请人发出接受NDA的通知,在120天内,申请人可以补交与安全相关的数据让FDA全面审评。
“FDA非常看重药物的风险获益比,以及试验方案的计划性、逻辑性,数据的科学性和方法学评价的合理性。中国公司如果能解释清晰,FDA还是会非常灵活地支持企业的做法。”肖申说。
为何要出海美国?
在小组讨论中,与会者认为,如果不打算把项目转让给其他美国公司,中国公司到美国开展临床试验需要建立整体规划。

从左至右为毛冬蕾、项安波、王俊、肖申、卢原。
石药集团临床事业部总裁兼非肿瘤领域CMO项安波博士认为,美国开展临床研究依旧有不可替代的优势。
首先,FDA拥有全球公认的科学监管审评实力,中国的产品一旦获得FDA批准,可能协助中国CDE开展审评决策。其次,美国具有多样化种族,有助于企业针对全球市场开发药物;第三,美国鼓励创新与科技,吸引全球临床研究具有丰富经验的专业人才;第四,美国是全球第一大医药市场,药品定价自由和商业化保险发达,企业能获得应有回报。
来凯医药副总裁、中国区临床开发负责人王俊补充说,全球生物技术公司之间的项目交易、联合开发发生得越来越频繁,已成为世界医药的主流。如果计划中美双报的话,在当地进行合作的企业,其临床数据对后续审评、融资都有较好的背书。
具体什么时机出海美国,项安波认为,各家公司的时间点可能不同。有些公司倾向于早期进入国际市场,在1期试验阶段就在澳洲开展,并利用澳洲的数据到美国申报。其他公司更看重在美国开展试验的完整性,1期会放在美国,继而与FDA在2期、3期整体沟通注册路径。对有计划在美国上市并且有实力开展中美双报的中国公司,越早到美国越好,可以获得更多美国多样性人群的数据。
王俊建议说,是否早期出海取决于公司的产品特性和策略。如果产品有创新性和新颖性,越早到美国等国际市场开展试验越有优势,可以与当地医院和医生建立联系,早期研究或为后续商业推广带来更多价值。
肖申说,FDA的审评要求越来越细致并不断变化,因此,早期与FDA讨论并获得其专家的指导有助于更好满足FDA的要求。他建议,早期在美国开展试验对产品全生命周期的数据评估特别是长期安全性评估十分重要。在海外积累的数据越多,与各国监管机构沟通交流越多,可以避免后续研究中的障碍。而晚期合作可以更全面、提供更有力的证据,为上市和产品推广奠定基础。另外,出海可以向国际合作伙伴学习,提高国内团队整体临床研究水平。
和誉医药临床项目高级总监卢原博士用一个出海的例子印证了肖申的观点。和誉的第一个治疗腱鞘巨细胞瘤的口服小分子pimicotinib(ABSK021)在中国、美国和欧洲同时开展全球2期临床试验。由于这类品种在美国获批较多,FDA、研究医院、患者用药的经验更多,和誉在与美国团队合作开展临床试验时收获的经验对国内研究和后续计划很有帮助。通过出海,和誉医药希望将其产品推向全球。
不同地区试验方案如何设计?
肖申认为,出海的临床试验方案设计多样化,涉及不同药物、适应症和疾病人群。监管部门更多考虑药物的安全性、试验终点、病人人群和药物剂量。卢原认为,考虑到不同疾病人群和药物剂量变化可能带来的复杂性,在临床前多开展一些功课是必要的,能为企业后续开发提供决策依据和信息。
具体在美国如何跟当地医院与PI合作?美国有约翰霍普金斯大学、杜克大学、纪念斯隆-凯特琳癌症中心等,当地医院的PI水平如何?如何招募患者进行给药和随访?
项安波说,这一流程跟国内大致相同。目前,美国大约1/4的研究中心设立在公立医院,3/4在私人医院。公立医院的研究费用较高但PI专业化程度高,病人资源广,而私人医院服务态度更好,也更愿意合作。每家医院的经验、每月招募病例数量也是重要考量。要与当地某一疾病领域最权威的医院和PI建立联系。通过拜访,详细告知项目的早期研究结果,未来试验计划。不过,能直接跟美国的PI沟通,需要强大的医学团队和专业知识。
在招募病人方面,美国有所不同。中国侧重于医生推荐病人,而美国更注重研究中心的推荐和招募广告。美国的招募广告创意十足,活灵活现。美国的受试者参与试验不完全是为了费用,希望为人类医学事业做贡献往往是其参与临床试验的目的。从试验费用来看,美国1期健康人试验每例大约需4万美元,而2期病人试验每例需要约9万美元。
王俊补充说,对于人生地不熟的中国公司,如果想打入美国临床界,可以先跟当地医学协会组织合作。例如来凯开发用于治疗卵巢癌的1类新药afuresertib(LAE002)在美国推进时,首先与美国妇科肿瘤学组(GOG)取得联系,该协会提供了全方位支持,包括协助来凯选择合适的研究中心开展试验,每两周与研究中心联系催促患者入组,定期更新入组情况,使项目进度可控。此外,GOG还会支持和参与FDA协商。对于生物技术公司来说,这种组织是除CRO以外的新选择。
在早期阶段,中国企业不可避免需要CRO。然而随着实力增强,可考虑在美国建立团队,从一两个医学博士开始来运营,无需大规模团队,许多医学信息可以由中国团队支持,并不需要花费太多费用。项安波提醒道,有时候也不能完全依赖CRO,需要自己的医学团队把控试验进度和质量。
出海如何明智选择CRO?
烨辉医药创始人华烨博士主持讨论了在出海过程中如何明智选择CRO。与会者认为,在美国开展临床试验,需要选择一家有经验、了解中国企业的可靠的CRO。

从左至右为华烨、周玉斌、朱海颖、陈丽娟、马东。
宜明昂科临床运营副总裁周于斌博士认为,在与CRO合作过程中,最关键的是CRO的企业文化是否与申办方相吻合,合作伙伴是否具有海外研究的经验和人员。
金赛药业临床运营副总裁朱海颖认为,选择CRO时,首先要考虑研究的特性,即是1期研究还是晚期试验,是否需要国际多中心合作。不同项目CRO的需求不同。其次,要评估CRO是否具有在相关领域开展试验的足够经验和成功案例。
康宁杰瑞注册负责人陈丽娟认为,1期研究更侧重于选择性价比较高的CRO,关键性研究需要考虑其经验和成功率。在全球合作上,需要与善于沟通和有相应经验的团队合作。
太美医疗科技高级副总裁马东认为选择CRO有三个考虑因素:一是项目情况,CRO的选择取决于项目的特定需求;二是公司战略,公司的国际化战略将影响合作伙伴的选择;第三是组织和管理能力,公司需建立有效的组织和管理能力,确保海外团队、研究医院能良好执行。上述这些决定在选择全球项目和管理方式时非常重要。
甲方乙方如何携手合作?
周玉斌说,为了更好地合作,需要制定一套完整的流程,制定工作计划,并设定预期目标。公司还需要建立评价系统筛选出最优秀的合作公司,将CRO视作自己内部的成员,协同管理,确保项目顺利进行。
陈丽娟说,一般而言,申办方会定期评估CRO的执行能力,跟进合同进度,提供反馈。同时建立内部协同团队,涵盖培训、诉讼和注册,这些部门会与CRO一起参与讨论研究细节和进度。另外,与CRO的沟通频率决定了项目进度。通常康宁杰瑞每两周沟通一次达成共识,提高工作效率。
马东介绍说,太美建立了一系列系统,包括CTMS、ETMF等被广泛用于管理临床研究项目。客户充分利用太美的系统确保数据的完整性和透明度,最大程度优化临床研究项目。最后,祝愿各家中国生物医药公司在出海美国的过程中能乘风破浪,顺利到达胜利的彼岸。

撰文|毛冬蕾
 产业资讯
医药魔方Info 2025-07-21
9
产业资讯
医药魔方Info 2025-07-21
9
 产业资讯
识林 2025-07-21
8
产业资讯
识林 2025-07-21
8
 产业资讯
研发客 2025-07-21
9
产业资讯
研发客 2025-07-21
9
 微信公众号
微信公众号 热门资讯
热门资讯 热点标签
热点标签