

 产业资讯
产业资讯
 研发客
研发客  2023-12-12
2023-12-12
 3669
3669
国家药监局坚持创新和国际化的总体方针不会变,将积极与各国药品监管机构继续开展国际交流与合作;由于MRCT设计和实施的难度加大,CDE需要与申请人详细讨论方案设计与实施。
第20届DIA日本年会的China Townhall上,国家药监局(NMPA)药品审评中心(CDE)统计与临床药理学部部长王骏博士介绍了国际多中心临床试验(MRCT)和ICH E17指导原则在中国的实施情况。

在中日两国药品监管和行业专家的共同推动下,中国药监局首次以China Townhall专场的形式在第20届DIA日本年会上登场。
从左至右为沈阳药科大学亦弘商学院研究员苏岭博士,日本PMDA国际事务副执行理事安田尚之,国家药监局中国食品药品国际交流中心主任董江萍,国家药监局药品审评中心统计与临床药理学部部长王骏博士,国家药监局食品药品审核查验中心核查中心检查一处检查员韩聪凡,武田中国副总裁、大中华区注册事务部负责人刘艳玮,泰格医药高级副总裁、首席医学官陈霞。
MRCT 成为全球研发趋势
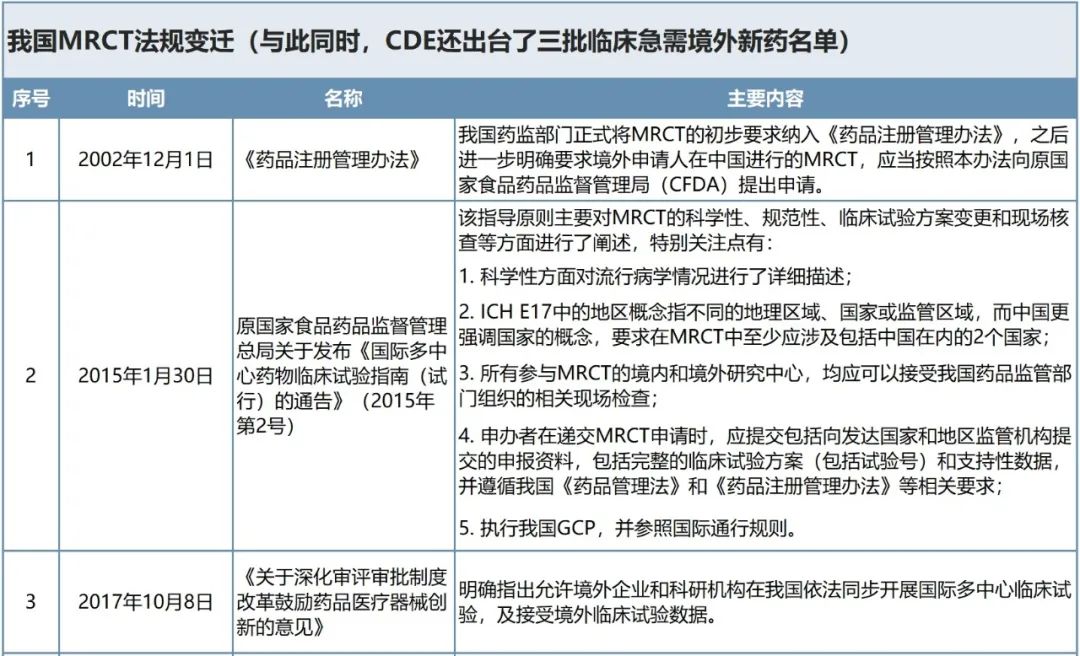
为了促进全球化药物同步研发,提高全球注册效率,ICH专家工作组通过ICH国际协调会议多次修订了E17“多地区临床试验计划与设计的总体原则”。据王骏介绍,NMPA在2017年11月16日最终采纳(第四阶段)ICH E17 MRCT的指导原则,并于2019年11月12日在中国正式施行。此后,中国企业开展MRCT用于全球注册的试验越来越多。
根据《中国新药注册临床试验进展年度报告》,2022 年新药临床试验中,MRCT占比达14.8%(292项),2021年新药临床试验中,MRCT达15.8%(321 项),虽然2022年因疫情原因比2021年略有下降,但对比近3年数据,MRCT占比逐年递增,2020年和2021年分别较上一年度增加21.6%和54.3%,2019 年和2020年分别为171项和208项。
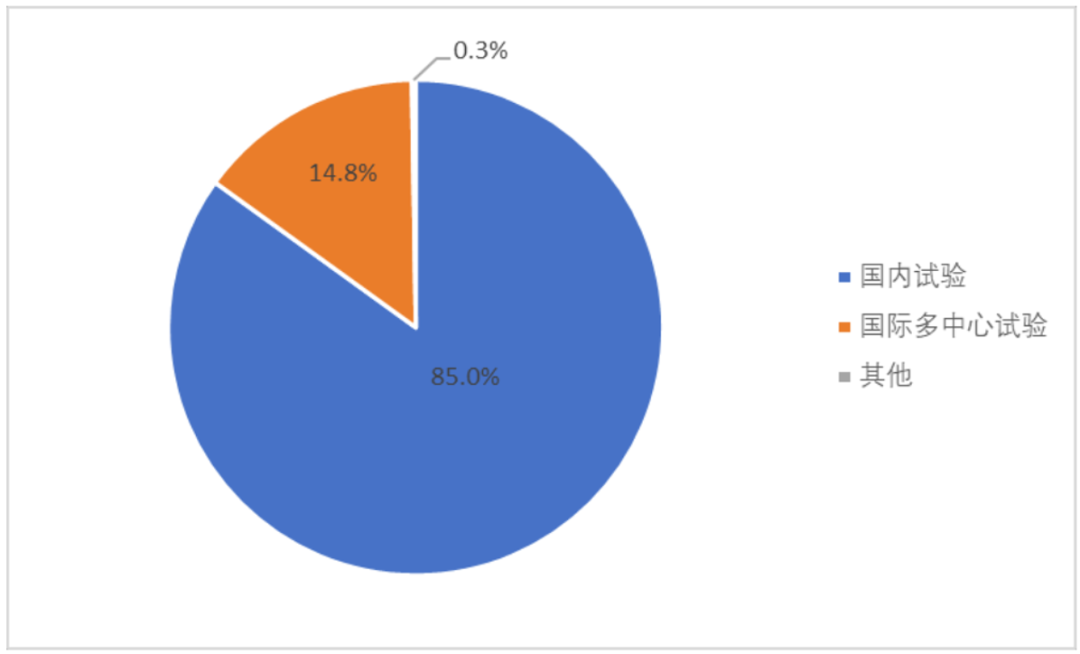
2022年新药临床试验范围分布 来源|CDE官网
王骏说,首先,MRCT带来的机遇与挑战并存。ICH E17只提供了技术要求,但缺少具体实操性的建议。CDE需要与申请人详细讨论方案设计与实施。而ICH E9(R1)等试验设计和分析方法的最新变化使MRCT的难度加大。为此,CDE开展了大量相关培训,建立工作机制,定期与学术界和产业界沟通。例如,CDE 与RDPAC的企业达成了样本量分配、试验一致性和稳定性评估、数据外推等共识。

王骏部长
其次,王骏介绍了如何建立完整的证据链。“我们需要全面、科学地评估区域研究结果。”为此,CDE提出区域获益-风险评估完整的证据链概念。完整的证据链由多个关键要素组成,如早期研究、足够的样本量、总体研究结果和全面的疗效评估。CDE正在考虑制定MRCT相关的整体科学评估框架。他认为,最重要的是区域结果与整体MRCT结果之间评价的统一和一致性。而一致性评价可考虑区域的医疗实践,患者的获益与风险。“CDE和申请人应朝着同一方向共同思考。”
第三,他介绍了审评过程中企业最关心的区域样本量分配。样本量的关键考虑因素是确保有足够的受试者可用于评估整体疗效,需评估各地区治疗效果的一致性,考虑总体样本量和地区样本量的分配。如:为何CDE要求中国人群15%~20%的样本量。
王骏说,CDE建议申请人参考ICH E17,平衡好按比例分配和平均分配。整体的分配方案根据区域定义、区域数量、各区域规模和各区域的疾病患病率来确定。“CDE不再笼统地要求区域之间互补,但要在充分了解医疗背景的基础上科学地考虑样本量。”他说。
他特别谈到了合并策略(pooling Strategy),常见问题包括企业使用合并策略的依据是什么。CDE认为,可以将合并区域的数据作为佐证。合并地区是指,当某些地区受试者在所研究的疾病和/或药物相关的内在和/或外在因素方面具有足够的相似性时,在试验计划阶段可将这些地区进行合并。
如果要以合并区域代替一个地区,则对早期研究提出更高要求,并要预先考虑地区间的差异。应预先规划合并策略,在试验设计时提供充分的数据支持,以便申请人与监管部门就合并策略的科学性达成一致。
“我们不应只关注人种差异等内在因素,而忽视医疗实践等外在因素可能造成的影响。”他提醒道,在MRCT设计时,要考虑在试验早期阶段探索内在/外在因素可能造成的影响,并进一步在确证性试验中收集相关信息用于对疗效的评估。
在小组研讨之前,沈阳药科大学亦弘商学院研究员苏岭博士收集了行业声音,提出合并策略中另一个问题:除中国临床数据外,其他地区的临床数据,如日本、东亚国家的数据是否可以用于支持中国药品注册?
《医药研发达人》主编高野哲臣认为,即使是在拥有14亿人口、56个民族的中国,使用东亚国家和地区的合并区域(pooled region)时,中国药监机构的要求也非常高,从制药企业希望以尽可能精简的临床数据包进行NDA申报的心态考虑,合并策略(pooling strategy)并没有那么吸引人/可实现。最后,大多数企业放弃了合并策略,为获得中国批准分别入组CDE所要求的中国人群样本量,为获得日本批准分别入组PMDA要求的日本人群样本量,现实情况是各国单独考虑各国的分别入组样本量。
王骏认为,CDE接受境外数据并不仅仅取决于数据来自哪里,同时还取决于数据的质量、数据是否能满足试验的目的、数据是否能证明药物的有效性或安全性。根据2020年10月12日国家药监局发布的《境外已上市境内未上市药品临床技术要求》(简称《技术要求》),用于治疗临床急需或罕见病的药品可以在中国豁免开展临床试验。
武田中国副总裁、大中华区注册事务部负责人刘艳玮介绍了CDE何时可以豁免临床试验的情形(见附表中《境外已上市境内未上市药品临床技术要求》的通告(2020年第29号),并分享了来自武田如何利用全球数据为中国产品上市提供支持的案例。
Revestive(Teduglutide,替度鲁肽),是一种治疗短肠综合征(SBS)的罕见病药物,早在2012年就在日本、美国和欧洲国家获批。由于中国患儿的临床急需,武田中国注册和研发团队与CDE沟通,提出日本人群等境外数据证实在安全性、有效性上无种族差异,且研究质量较高。基于这些考量,CDE初步同意利用境外数据支持上市,免除在中国进行临床研究。刘艳玮说:“CDE要求我们开展上市后研究,而患儿则提前3~5年获得了该药。”
过去3年,武田共有5个药物运用日本及其他海外数据在中国免临床获批。“CDE的灵活和开明,为武田后续制定临床战略带来新的思考,我们会越来越多基于日本人群和境外数据到中国申报。”刘艳玮说。
泰格医药高级副总裁、首席医学官陈霞介绍了我国《药品注册管理办法》中新药的5种注册分类。她认为,除了1类创新药以外,某些改良药有时也可以考虑全球同步研发的情况。尽管主流监管机构都遵循科学监管原则,但对于某些问题,不同监管机构有不同的主张,如某些适应症能否依据替代终点批准,或对改良药的审评要求等。她建议企业在制定涉及中国的全球研发计划时,尽早与CDE沟通,有利于CDE了解企业的想法。
王骏阐述了CDE关于一致性评估的思考。“一旦发现不一致性,CDE建议采用多种方法开展一致性评价,包括描述性方法和推论性方法。而非告诉我们那是偶然的,或只是提供简单的文献回顾。”
早期临床研究可以为MRCT中确证性研究的设计提供支持,也能为新药上市提供进一步支持。CDE鼓励申办方在开展全球确证性MRCT之前,尽可能在中国开展早期研究,并将中国人的数据纳入。
日本PMDA国际事务副执行理事安田尚之总结认为, MRCT将成为全球药品研发和监管的趋势。如果跨国大药厂能尽早把中国纳入MRCT的一部分,这将是中国贡献数据最高效的方式。
中国监管全面国际化
在国际合作方面,苏岭提问:国家药监局未来在国际同步研发、试验设计、数据申报以及审批审评上,与全球监管机构有哪些合作?

董江萍主任
国家药监局中国食品药品国际交流中心主任董江萍说:“NMAP一贯高度重视药品监管领域的国际交流与合作,深入参与药品监管治理,与国际社会的合作交流持续深化。”她详细介绍了中国抗新冠疫情的国际合作、参与国际规则的制定、开展传统药国际协调以及对标国际加强能力建设。
“NMPA坚持创新和国际化合作的总体方针不会变。下一步,NMPA将积极与各国监管机构开展国际合作。包括加入PIC/S,深入参与ICH的指南制定和实施,在GCP和GMP核查方面全面与国际交流合作。我们相信,世界越来越需要中国。与此同时,中国也积极融入世界。只有建立了强大的监管体系才能促进制药产业的发展,成为制药强国,把更多满足临床价值的药品带到全球。”董江萍说。
据董江萍介绍,NMPA共向ICH 40个议题工作组选派了70名专家参与ICH指导原则的制修订工作,中国已采纳实施全部ICH指导原则,同时也在和世卫组织、ICMRA等国际机构积极合作。2023年2月16日,国家药监局副局长徐景和当选第27届全球医疗器械法规协调会(GHWP)主席。

韩聪凡检查员
来自NMPA食品药品审核查验中心核查中心检查一处检查员韩聪凡介绍了中国药监局近年来稽查的情况,如何从监管和稽查的视角持续提高中国临床试验的质量。

China Town Hall结束后讲者和与会人员合影。

感谢国家药监局中国食品药品国际交流中心、药品审评中心、食品药品审核查验中心、日本DIA办公室、中国DIA办公室、China Townhall工作小组对本文的大力协助!
 产业资讯
药融圈 2025-07-07
111
产业资讯
药融圈 2025-07-07
111
 产业资讯
医药魔方Pro 2025-07-07
115
产业资讯
医药魔方Pro 2025-07-07
115
 产业资讯
药研网 2025-07-07
116
产业资讯
药研网 2025-07-07
116
 微信公众号
微信公众号 热门资讯
热门资讯 热点标签
热点标签