

 产业资讯
产业资讯
 中国医药创新促进会
中国医药创新促进会  2025-06-14
2025-06-14
 1687
1687
引言
当全球生物医药产业驶入深水区,创新药“出海”已不仅是企业扩张的必选项,更是中国医药产业升级的战略支点。日本作为全球第三大医药市场,与中国药企在靶点开发、成本控制方面优势互补,成为中国创新药企出海的重要一站。
中国医药创新促进会始终致力于深化中日医药产业合作,并在医药创新领域取得了一系列实质性成果。为全面助力中国药监、生物技术公司、投资界深入了解日本药品监管法规、申报流程、沟通交流以及生产和检查等相关事宜,我会联合研发客、上海市生物医药科技产业促进中心以及泰格医药,共同开设“出海日本”专栏,特邀日本法规监管领域的资深专家发布专业性文章。撰稿人包括著名的药品开发及监管专家高野哲臣先生、东内祥浩先生和毛冬蕾女士。同时研发客主编毛冬蕾女士还将对日本政府、学术界以及中日两国业内专家进行访谈,共同探讨开发及监管热门话题。
中国药促会中日医药合作交流
联系人:马明尧
电话:13520846026
邮箱:mamy@phirda.com
撰文|医药研发达人主编 高野哲臣(t2T Healthcare股份公司总裁兼首席执行官)
翻译|项安波(石药集团) 董方(东方伊诺医疗科技)
中文版翻译负责人|医药研发达人主编 高野哲臣
•受美国影响,日本于1985年开始导入临床试验伦理委员会(IRB)应在每家研究机构分别设立制度,即1990年10月施行的旧GCP明确规定IRB原则上应在每家研究机构分别设立,直至2008年3月GCP省令修订才被废除该规定。然而此后IRB中心化并未推进,目前日本仍有约1300个临床试验IRB,中心IRB在许多研究机构普及程度不充分。
•在IRB中心化方面欧美与日本存在差异。欧盟采用去机构化的中心IRB审查体制,遵循“一个成员国,一个意见”原则;美国正加速向商业IRB高度集中方向发展;而日本的IRB中心化进展缓慢。
•为解决“药品错失和药品上市延迟”问题,日本正加速临床试验IRB的变革,不仅推动中心IRB,更向单一IRB迈进。
本系列A《日本的临床试验与药品市场》第五篇,由高野向大家介绍日本的IRB的历史背景、发展过程、现状,以及正在发生的变革与未来走向。
需要注意的是,本文多处亦包含笔者的主观看法,敬请读者留意。
01
日本临床试验IRB的历史
全球各国的伦理委员会(IRB)之起源可以追溯到20世纪60年代的美国。在美国构想IRB制度时,临床研究大多在单一的研究机构内进行。1974年,美国制定的《国家研究法》(National Research Act)规定,接受联邦资助的研究机构必须设立“机构内部审查委员会(Institutional Review Board,IRB)”,对临床研究计划进行事前审查。(来源:田代志门《医疗与社会》28卷1期,第79-91页,2018年等)
日本关于药物临床试验实施的标准(旧GCP)于1985年12月公布草案,随后于1989年10月作为厚生省药务局局长通知正式公布,并于一年后的1990年10月开始实施。该旧版GCP沿用了“Institutional(机构内)”的概念,明确规定IRB原则上应在每家研究机构分别设立。
随后,在1996年5月,ICH-GCP达成共识并进入第4阶段,其并未规定各研究机构必须单独设立IRB。
为了落实ICH-GCP的要求,日本于1997年3月公布了对应的GCP省令(新GCP),并于1998年4月全面实施。然而,尽管该GCP省令基本上反映了ICH-GCP的内容,但由于是在延续1990年10月施行的局长通知(旧GCP)的基础上制定的,因此仍明确规定每家研究机构原则上必须设立IRB。
这一原则性规定,直到GCP省令公布11年后的2008年3月GCP省令修订时才被废除。根据PMDA公开信息(https://www.pmda.go.jp/review-services/trials/0008.html),截至2025年4月30日,日本依然存在约1,300个临床试验IRB。这表明,即使在2008年3月以后,日本的IRB数量并未出现明显减少。笔者认为,在2008年3月以前就已设立院内IRB的医疗机构,向“废除院内IRB → 使用院外IRB”方向转变的趋势依旧缓慢。(参见第8期图1)
02
欧美临床试验呈现IRB中心化趋势
在欧洲,伦理委员会(IRB)多数被定位为公立机构,数量相对较少。例如,法国有39个IRB,英国为87个。相较于历史上由各研究机构自行设立IRB的美国与日本,欧洲的IRB数量显著偏少。此外,在各欧盟成员国中,只要获得该国某一家IRB批准和其监管部门许可,即可启动临床试验。
此外,欧洲在《临床试验指令》(Clinical Trials Directive 2001/20/EC)中还明确规定了“一个成员国,一个意见”的原则。也就是说,欧洲去机构化的中心审查体制已成为基本前提。此后,该指令由《临床试验条例》(Clinical Trials Regulation (EU) No 536/2014,于2022年1月底生效)所取代。尽管在《临床试验条例》中对IRB相关规定进行了若干重要修改,但“一个成员国,一个意见”的基本原则仍然得以保留。
另一方面,为了在美国推进中心IRB(Central IRB),FDA于2006年3月发布了《行业指导原则:在多中心临床试验中使用中心IRB审查流程》(Guidance for Industry: Using a Centralized IRB Review Process in Multicenter Clinical Trials)。不过,其不具法律约束力,仅作为推荐性意见。此外,美国国立卫生研究院(NIH)于2016年发布了指南,要求由NIH资助的临床试验必须采用单一IRB(Single IRB)进行审查。
截至2023年,美国约有2,300个IRB分布在1,800家研究机构或组织中。其中,大学IRB占比过半(1,284个,占56%),其次为研究机构(553个,24%)、民间组织(229个,10%)、政府机构(190个,8%)以及独立IRB(47个,2%)。
在美国,受FDA监管的IND临床试验(每年约2,000项左右)中,由独立IRB(包括非营利IRB及商业IRB)进行审查的占比,从2012年的25%上升至2021年的48%,首次超过大学IRB跃居首位。值得注意的是,独立IRB的数量本身因整合而有所减少,目前呈现由两家商业IRB(WCG公司与Advarra公司)寡头垄断的格局, 2021年的IND临床试验中,仅这2家公司承担的IRB审查数就占了独立IRB的92%。这两家公司的IRB会议皆为每日召开,IRB委员人数超过150人。据称,WCG公司2020年仅IRB相关业务与咨询服务的毛利润就分别超过1亿美元。
(本章节资料来源:2023年5月《国内外临床试验相关环境最新动向调查研究》,令和4年度 总括·分担研究报告书,佐藤晓洋等;2024年3月21日《第九届加强药品研发能力和确保稳定供应的监管研讨会》资料3)
03
尚未推进中心化的日本临床试验IRB
如前所述,在欧洲,IRB自设立之初即为中心化体系,并已在现行法律框架下,仅由公立中心IRB负责运营。而美国虽最初以研究机构IRB为主,但自2020年前后迅速推进中心化,目前正加速向商业IRB高度集中的方向发展。
然而,在日本,虽然在2008年3月通过修订GCP省令,废除了每家研究机构必须设立IRB的原则性义务,但其后IRB的中心化并未显著推进。尽管在部分诊所及国立医院机构中,中心IRB的使用已有所发展,但在大学附属医院及公立医院等研究机构中,中心IRB的普及程度仍不充分。
需要指出的是,中心IRB所具备的优点,其实正是解决了各研究机构分别设立IRB所存在的不利之处:如在申报前各研究机构需进行大量准备工作及等待排队审查,部分研究机构导致IRB批准可能耗时数月;且各IRB对申请资料的要求不尽相同,给申办者(包括CRO)带来较大负担;最终导致临床试验启动周期延长,研究机构与申办者双方的行政工作量剧增,进而抬高成本;此外,在IRB审查的一贯性与质量保障方面也存在一定担忧。
为了解决自2022年左右以来在日本凸显的“药品错失和药品上市延迟”问题,日本厚生劳动省(MHLW)于2023年7月至2024年3月设置了《加强药品研发能力和确保稳定供应的监管研讨会》(监管研讨会),并于2024年4月形成了报告。关于包括中心IRB推广在内的临床试验效率提升(即引入“临床试验生态系统”)议题,在2024年3月21日举行的第九届监管研讨会中进行了重点讨论(参见图1)。
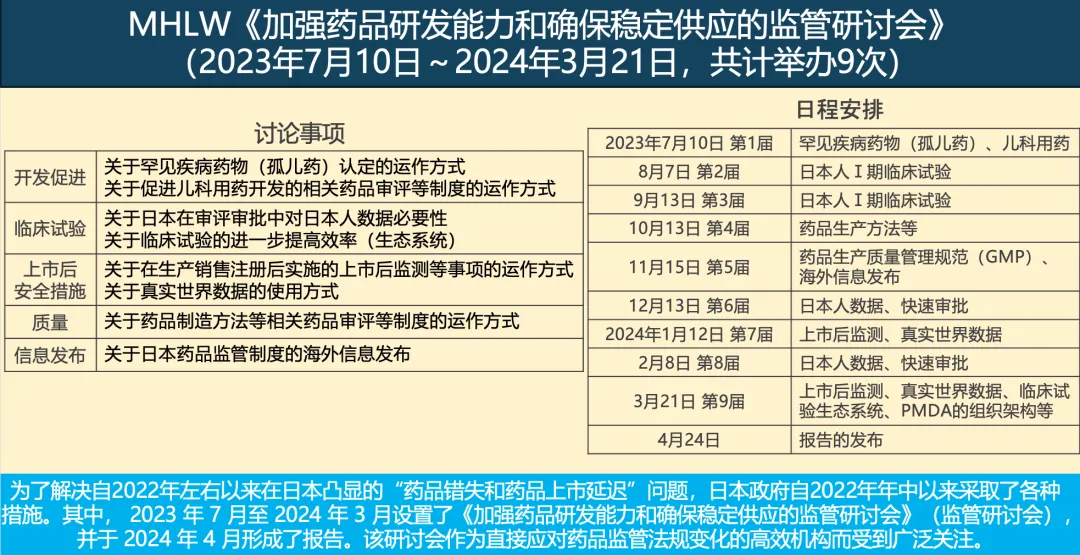
图1. MHLW《加强药品研发能力和确保稳定供应的监管研讨会》(2023年7月10日~2024年3月21日,共计举办9次)
(引自关西药品协会,MHLW吉田易范大臣官房审议官特别演讲会,大阪,2024年5月29日,部分改编)
在第九届监管研讨会上,有专家指出,在日本中心IRB普及不力的原因在于,研究机构难以享受到中心IRB带来的好处,反而面临更多弊端(见图2)。笔者认为,图2中所列的诸多原因,与中国的中心IRB未能普及的原因存在高度共性。

图2. 日本中心IRB未能普及的原因(引自:
MHLW第九届《加强药品研发能力和确保稳定供应的监管研讨会》,资料3,东京,2024年3月21日)
04
并非止步于中心化,日本临床试验IRB正加速迈向单一IRB
由于中心IRB具备减轻事务工作量、降低成本、加快临床试验启动速度、确保审查一贯性与质量等多重优势,第九届监管研讨会结论提出了鼓励采用中心IRB的方针,且“原则上应由中心IRB进行审查”这一点将以书面形式加以明确。目前,MHLW、药品医疗器械综合机构(PMDA)以及制药界正听取医疗相关人士的意见,推进相关文件的制定与发布工作。
特别值得关注的是,由内阁官房健康·医疗战略室负责雜務工作的“提升药品研发能力,将最新药品快速送达国民的构想会议”在2024年5月22日公布的中期总结中,提出了要在日本的多中心临床试验中,将中心IRB的最终进化形态——“每项试验方案(Protocol)仅由单一IRB审查(Single IRB)”作为原则,并致力于解决实施该原则所面临的制度与流程障碍。从2024年开始,日本在临床试验IRB的制度改革上,将不仅仅是推动中心IRB,而是直接向单一IRB迈出关键一步。
笔者注释:
单一IRB (Single IRB)是中心IRB模式中最理想且最高效的形式,即单一IRB的定义与中心IRB(Central IRB)有所不同。例如,在日本部分诊所及国立医院机构中,中心IRB的使用已有所发展,这有助于减少整个临床试验中的IRB数量,但尚未开始建立全国单一IRB。也就是说,日本与欧洲的情况不同。另一方面,中国的状况与日本相似,虽然各市和省可能设有中心IRB,但并非覆盖中国所有研究机构的"单一IRB(按单一试验方案)",尚未建立参与全国的多中心试验或MRCT的全国单一IRB。(高野哲臣)
作为这一趋势的佐证,目前MHLW正在讨论的《2025年GCP省令等的主要修订内容》中,“单一IRB原则化”已被列为首要议题(见图3)。

图3. 2025年 J-GCP (GCP省令) 等的主要修订内容(正在讨论中的草案)
(引自:MHLW审评管理课,2024年度临床试验生态系统导入推进事业成果报告会,东京,2025年3月24日,部分改编)
目前,ICH-GCP(E6)正在进行全面修订,其最新版本E6(R3)的原则(Principles)及附件1(Annex 1)已于2025年1月6日达到第4阶段,而E6(R3)的附件2(Annex 2)也已于2024年11月至2025年3月进行公开征求意见。
为反映E6(R3)内容,J-GCP(GCP省令)预计将在2025 - 2026年左右进行全面修订。届时,将利用此次修订机会,针对IRB,除推进上述单一IRB外,还期望推进接受英文资料、通过整理IRB审议事项等实现程序合理化、提高审查效率、推进临床试验电子化与数字化转型(DX)、集约化与高效化处理事务性工作,以及随之带来的研究机构和临床试验申办者双方的成本削减等内容。
通过IRB的集约化,不仅可以加快临床试验启动速度、提高效率,还能够提供适应复杂化、多样化临床试验的高质量审查功能。这些正是近年来日本临床试验申办者对IRB系统的核心期待——如今,这些愿景正逐步成为现实。
综上所述,本文回顾了“日本的临床试验IRB”从历史沿革、现状分析到未来制度改革的全貌。可以明确预见的是:2025至2026年,日本的临床试验IRB体系将发生重大变革。
下一篇(系列A第六篇)将介绍:关于“日本药品监管部门应对药品错失和药品上市延迟的措施示例”中的“日本临床试验费用的计算方法的合理化与透明化——引入基于公平市场价值(Fair Market Value)的基准型成本计算机制”等内容(参见第3期图4之第6项内容)。敬请期待。
下期预告 Next Preview
下期(第12期)预计于2025年6月下旬出版,内容为东内祥浩先生撰写的系列B《日本的监管制度及其实际情况》中的第五篇,主题为《日本的临床试验申报》。高野哲臣先生撰写的系列A《日本的临床试验和药品市场》第六篇《日本的临床试验费用计算方法》预计将于2025年7月上旬出版。
 产业资讯
瞪羚社 2026-06-18
406
产业资讯
瞪羚社 2026-06-18
406
 产业资讯
深蓝观 2026-06-18
420
产业资讯
深蓝观 2026-06-18
420
 产业资讯
研发客 2026-06-18
463
产业资讯
研发客 2026-06-18
463
 热门资讯
热门资讯 微信公众号
微信公众号