

 产业资讯
产业资讯
 同写意
同写意  2026-03-28
2026-03-28
 1637
1637
中国正迅速崛起为全球药物研发的主要中心。
一组直观的数据是,从2015年到2024年,全球药物研发项目数量从约10400个增至19000个,增幅近一倍。其间,美国项目从约5000个增至7000多个,但全球占比从48%降至37%;而中国从830个增至6000多个,成为全球增长的主引擎。
随着中国创新能力与地位的跃升,如何将其日益增长的研发实力纳入监管框架,以确保药物能够及时、公平地惠及全球患者,正成为一项关键命题。
3月26日,美国FDA药物评估与研究中心(CDER)前主任、曾长期担任肿瘤卓越中心主任的理查德·帕兹杜尔(Richard Pazdur)在《美国医学会杂志》(JAMA)联合发表了一篇文章,揭示了药物创新地理格局的深刻变迁。
文章指出,中国已崛起为药物研发的主要中心;面对这一变化,监管机构应主动作为,通过加强多区域临床试验协调、完善桥接试验标准、推动数据共享等举措,缩小因研发活动地理转移而可能出现的创新疗法可及性差距,确保全球患者能够同步受益于生物医学进步。
TONACEA
01
全球创新格局的重塑
无论是BD交易、跨国投资,还是近期获批的“首创性”(FIC)药物,均表明中国药物研发管线正日益囊括创新与变革性疗法,已超越早期以开发后续产品为主的阶段。
在ADC、双特异性抗体和细胞疗法等领域,中国在规模上已与美国比肩甚至实现超越,并在部分领域展现出独特的创新优势。
波士顿咨询公司(BCG)近期发布的报告进一步揭示,自2021年起,中国每年新进入临床阶段的创新药新分子(NME)数量已超过美国,并在随后几年持续扩大领先优势。
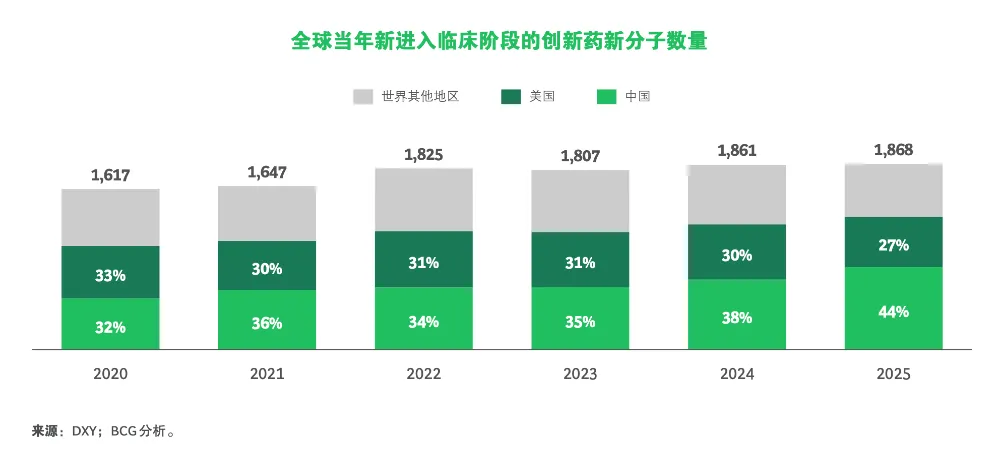
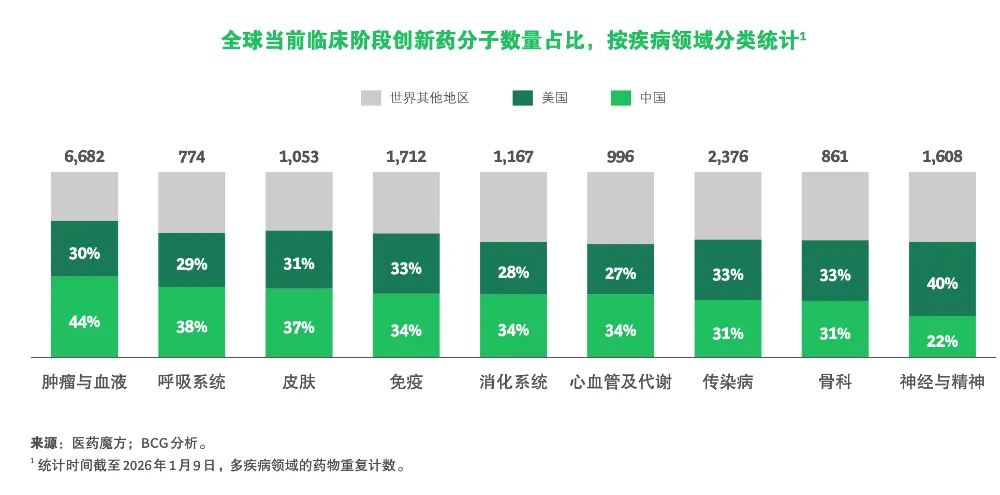
更值得关注的是,这种“数量”的增长正从单一赛道的高度集中,演进为数量与广度并进的结构性扩张——在呼吸系统、皮肤、免疫、消化系统与心血管等全球研发热点领域,中国当前处于临床阶段的创新药分子占比已超过美国。
十五年前,中国制药业主要聚焦于原料药生产以及为本国市场供应仿制药制剂。彼时,对临床试验实施和数据质量的担忧,也在一定程度上限制了国际市场对中国研发疗法的接受度。
过去十年,中国开展的临床试验质量显著提升,中国制药企业也逐步从仿制药生产,迈向为国际市场开发新型疗法的新阶段。
美国药品研究与制造商协会(PhRMA)首席执行官斯蒂芬·乌布尔(Stephen Ubl)在2026年2月的一次行业活动中指出,"中国开展I期临床试验的速度比美国快50%,成本低40%"。
他补充道:“过去关于中国开发'me-too'药物的旧说法,已被新现实所取代——中国现已成为免疫疗法、癌症疗法和自身免疫性疾病等领域的创新领导者。”
Pazdur在文章中总结,若干结构性因素共同促成了中国成为研究、开发和制造中心。研究人员可获得高质量、低成本的临床前研究资源,拥有规模庞大且能力日益增强的临床试验基础设施,并受益于旨在加快审评和商业化的监管改革。
此外,越来越多的中国学生在国内完成本科教育后赴海外攻读研究生学位,随后回国发展。许多曾在西方制药公司或美国政府机构任职的管理层,开始创立Biotech公司,带来了药物开发、监管和商业运营方面的丰富经验。
持续不断的公共和私人投资,以及中国生物技术公司与跨国制药公司之间日益深化的合作伙伴关系,已将中国本土项目纳入全球研发管线。
在监管改革方面,例如为在研究者发起的试验(IIT)中,进行研究的细胞和基因疗法开辟并行的商业化路径,也有助于缩短创新周期,从而优化现有疗法并催生新药。
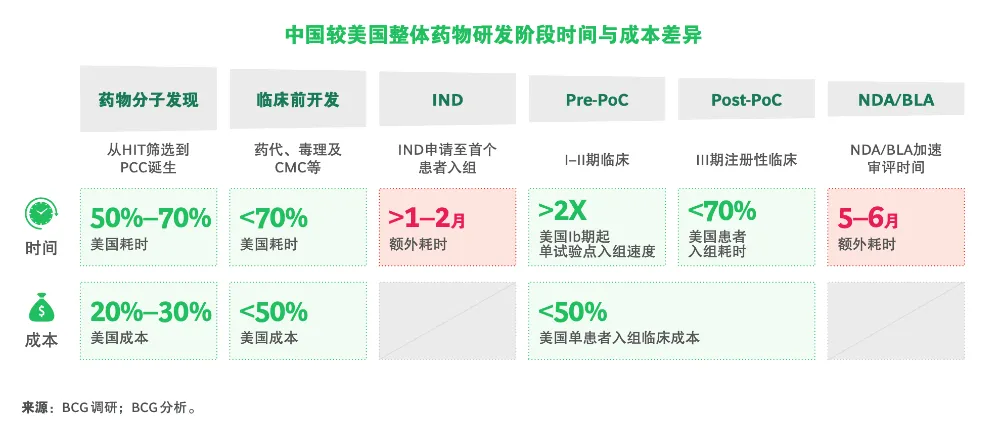
BCG报告则系统性地解构了中国医药创新背后的“效率飞轮”——“多、快、省”三者叠加形成的体系化能力:
“快”与“省”:从靶点确定到新药上市申请的端到端流程中,中国已成为全球医药创新的“速度极”与“成本洼地”。
在药物分子发现阶段,其成本仅为美国的20%-30%,临床前开发花费也较美国节省至少一半。同样的优势延伸至临床开发阶段,依托庞大的患者基数与更为集中的患者及医院网络,中国以入组速度快著称。
“快”与“省”的底层逻辑:BCG将其归结为“理解—试错—执行”的闭环。
中国团队凭借多年技术与经验积累,能够更快理解全球前沿机制,形成更聪明的快速跟进能力;依托工程师红利和低成本迭代条件,能够更快完成“设计—测试—学习”闭环;完备的合同研究、开发和生产组织(CRDMO)生态则将高频迭代落实到端到端交付上。
TONACEA
02
全球监管体系的影响与应对
Pazdur认为,这种研发活动地理格局的重塑,对患者、监管机构和政策制定者均产生了深远影响。
全球研发能力的扩展可能增加疗法的供给,加剧商业竞争,从而有望改善药物可及性并抑制价格。与此同时,随着首次人体试验越来越多地在中国启动,美国患者可能面临在研疗法获取延迟的问题,美国临床医生参与早期研究的机会也可能相应减少。
当然,其影响远不止于临床试验。制造与供应链地理集中度的提高,使得药品供应更易受到地缘政治紧张、贸易争端或监管碎片化的冲击。
基于此,文章指出,随着研发活动地理分布的变化,围绕既往创新格局设计的监管体系也需要相应调整。促进和鼓励多区域临床试验——即跨越多个地理区域开展的大规模试验——对于确保支持新疗法的证据具有跨人群的普适性,并能够支持各区域的同步获批至关重要。
在Pazdur看来,为减少全球审批延迟和患者获取药物的等待时间,申办方应及早识别在中国开展的创新产品早期试验,并在多区域框架内设计后续的注册研究。
同时,监管机构必须为这样一种环境做好准备:即某些能为患者带来显著益处的疗法可能主要,甚至完全是在中国进行研究的。这在罕见病领域尤为棘手,因为患者群体较小,在各区域重复试验可能不切实际或在伦理上存在困难。
现实的情况是,无论疾病发病率高低,当为具有显著疗效的新药提供关键数据主要来自一个人口相对同质化的地区时,监管机构往往面临着复杂的权衡:是接受非本国产生的临床数据,要求进行针对性的桥接试验,还是坚持开展重复中国已进行试验的额外确证性试验。
对此,文章认为,监管决策需在潜在获益与潜在风险之间取得平衡。能带来有意义的临床获益(如总生存期OS改善)的疗法,可能需要在接受来自其他国家的数据提交方面给予更大的监管灵活性。
桥接试验通常指为评估药物在不同地区是否存在药代动力学、药效动力学、安全性或有效性差异而开展的小规模研究。当存在生物学上合理的显著种族或地区差异时,这类试验不失为一种合理选择。明确桥接试验的适用条件,以及其应在申报前还是申报后进行,对于保障患者及时用药、维护临床试验数据适用性的公信力,具有重要意义。
总之,随着中国研发活动的扩展,监管机构应主动采取措施,缩小创新疗法可及性方面可能出现的差距。在试验设计、检查标准和数据共享实践方面尽早达成共识,有助于促进各区域的同步申报,并缩短从验证安全有效性到全球患者获得药物之间的时间差。
整体来看,中国在全球药物研发中日益增长的参与度,已成为不可逆转的趋势。
面对这一深刻变革,与其构筑跨境创新的壁垒,不如以促进高效开发与监管合作为方向,让竞争与合作在完善的规则框架下并行不悖。唯有如此,生物医学科学的进步才能真正跨越国界,及时转化为惠及全球患者的切实福祉。
 产业资讯
synbio深波 2026-06-18
372
产业资讯
synbio深波 2026-06-18
372
 产业资讯
医麦创新药 2026-06-18
443
产业资讯
医麦创新药 2026-06-18
443
 产业资讯
动脉网 2026-06-18
418
产业资讯
动脉网 2026-06-18
418
 热门资讯
热门资讯 微信公众号
微信公众号