

 研发追踪
研发追踪
 药明康德
药明康德  2022-11-28
2022-11-28
 3119
3119
argenx今日宣布美国FDA接受其efgartigimod皮下注射制剂,用于治疗全身性重症肌无力(gMG)成人患者的生物制品许可申请(BLA),并授予优先审评资格。FDA预计于2023年3月20日完成审查。
重症肌无力是一种罕见的慢性自身免疫性疾病。患者体内的IgG抗体会破坏神经和肌肉之间的沟通,引起虚弱和可能危及生命的肌无力。超过85%的患者在发病后18个月内进展为全身性重症肌无力,进而导致极度疲劳和面部表情、言语、吞咽和活动困难。
Efgartigimod是一款靶向Fc受体(FcRn)的“first-in-class”疗法。其静脉输注(IV)制剂Vyvgart(efgartigimod alfa-fcab)在去年12月获批,成为首个FDA批准的FcRn阻断剂。Efgartigimod可减少致病性IgG抗体,阻断IgG再循环过程。FcRn受体的作用是防止IgG的降解,因此通过防止IgG与FcRn的结合,能够导致介导自身免疫性疾病的IgG抗体更快耗竭,从而减轻疾病症状。再鼎医药已与argenx公司达成合作,获得这款创新疗法在大中华区的开发和商业化权益。
Efgartigimod皮下注射制剂包含重组人透明质酸酶PH20(rHuPH20)。rHuPH20由Halozyme Therapeutics公司开发,它可以降解体内的透明质酸,以帮助皮下注射药物的渗透和吸收,为患者提供额外的治疗选择。
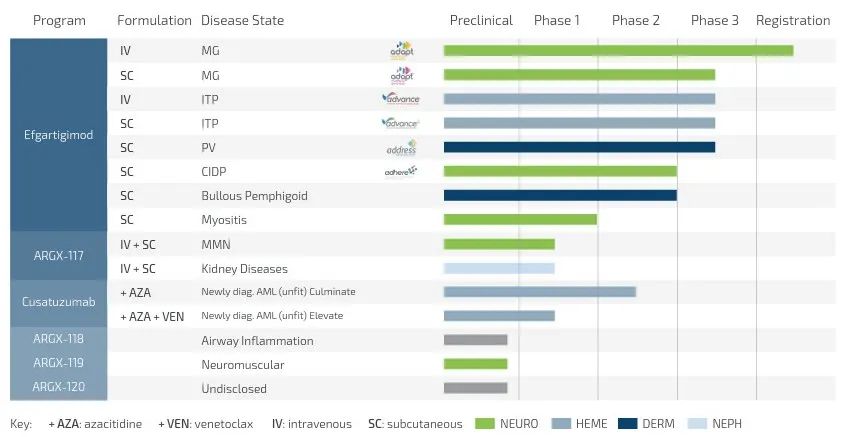
▲argenx公司研发管线(图片来源:argenx公司官网)
这次申请递交是基于ADAPT-SC临床3期试验的结果。该试验在110位成人gMG患者中评估了efgartigimod皮下注射制剂与静脉输注制剂相比的药效学作用。试验达到了非劣效性标准的主要终点(p<0.0001),皮下注射制剂组第29天的平均总IgG水平相对基线降低66.4%,而静脉输注制剂组为62.2%。此外,试验还达到其他关键次要终点,与在静脉输注制剂3期临床试验中观察到的临床疗效结果一致,包括重症肌无力日常生活活动(MG-ADL)和定量重症肌无力(QMG)等评分的改善。其安全性特征也与以往研究一致,通常耐受性良好,最常见的不良事件是注射部位反应(ISR)。
“FDA接受我们BLA是完成我们拓展gMG患者疗法以协助每位病患经历这项衰弱性疾病的重要一步。我们对于gMG患者能够有机会通过多种方式接受不同剂量的治疗感到兴奋,”argenx的首席运营官Keith Woods先生说道,“有了确切的PDUFA日期,我们正在准备推出我们第二项商业化产品,我们期待有机会为gMG患者再推出另一项治疗选项。”
 研发追踪
DailyBio 2025-05-12
96
研发追踪
DailyBio 2025-05-12
96
 研发追踪
UmabsDB 2025-05-12
98
研发追踪
UmabsDB 2025-05-12
98
 研发追踪
药明康德 2025-05-12
100
研发追踪
药明康德 2025-05-12
100
 微信公众号
微信公众号 热门资讯
热门资讯 热点标签
热点标签