

 产业资讯
产业资讯
 同写意
同写意  2023-06-01
2023-06-01
 2039
2039
当 Exondys 51 (Eteplirsen)2016 年被加速批准用于治疗杜氏肌营养不良症(DMD)时,并没有证据表明该药物确实能减缓这种疾病。Exondys 51加速批准引发了巨大争议,当时两名 FDA 审查小组成员辞职以示抗议。7年后,Exondys 51的制造商 Sarepta Therapeutics Inc.仍然没有完成批准后研究(post-approval study),提供确凿的数据证明其有效性以及安全性。迄今为止,Sarepta Therapeutics Inc. 已经从 Exondys 51 和两种相关药物中获得了超过 25 亿美元的销售额。这三者都通过FDA的加速批准,允许公司在患者几乎没有治疗选择的情况下销售药物。

图片来源:Exondys 51.com
Sarepta在Exondys上躺赚四年之后,才于2020年意兴阑珊地开始了批准后的验证性研究,而且计划到2024年才能完成。也就是说,如果2024年的研究数据证明,Exondys没有实际临床益处,或者其利益无法抵消风险的话,即便FDA以雷霆万钧的最快速度勒令其退市,Sarepta也已经无中生有地获取了八年的销售利润。
这种“知其也许不可而为之“的做法在业内并不罕见。一项研究发现,有19种得到FDA加速批准的药物,截止今年4月,其批准后验证研究处于延迟状态,包括超过官方截止日期的研究以及落后于计划的试验。“只要没证明无效,就是有效”的逻辑可以保障这些药物继续在市场上流通销售。该分析还发现,另外7种加速批准药物的批准后研究落后于计划,但由于这些公司已向FDA提交了一些数据,因此未被归类为延迟。严格地说,26种加速批准药物的批准后研究没有能够按照当初的时间计划进行。
“只要我不尴尬,尴尬的就是别人”。Sarepta近日进行了一场关于他们最新的杜氏肌营养不良症基因疗法的FDA咨询委员会(Adcomm),对其产品SRP-9001进行讨论表决。尽管最终以8:6的投票数艰险获得顾问小组成员支持,但两名持反对意见的成员指出,Sarepta之前的三款加速批准产品,没有一个完成了批准后研究。被扒出黑历史,也证明了Sarepta在完成批准后研究的态度问题。
1 加速批准途径如何运作?
加速批准(accelerated approval)最初是为应对 HIV危机而于 1992 年由FDA创建的制度,它的核心是允许使用“替代终点(surrogate endpoint)”作为标准来加快药物批准,而不是像传统 FDA 批准所要求的那样,必须通过直接衡量临床益处(clinical benefit)作为临床终点。替代终点是已知或可能预测临床益处,但不直接衡量临床益处的替代指标。例如,提交加速批准的药物的临床益处,可能通过实验室测量或放射成像,而不是其延长寿命的能力来确定。Exondys 获得批准的数据显示,它略微提高了肌营养不良症患者所缺失的肌肉蛋白抗肌萎缩蛋白(dystrophin)的水平。Dystrophin就是以生物标志物(biomarker)的方式证明了药物的临床替代终点。
一旦通过加速批准途径获得批准,制造商必须进一步研究(即post-approval study, 批准后研究)以确认该药物的临床益处。但如果试验未能验证临床益处,或者没有足够证据来证实临床益处可以抵消与药物相关的风险,FDA则可能会撤回药物的批准。
2 加速批准:药物上市的终南捷径
加速批准不仅加速了药物的上市,就连加速批准本身也在“提速”之中。1992年,加速批准只是孤例;18年后,当年的一条好汉迅速扩增到了47。即便2020年的峰值不具完全代表性,2022年也见证了15款通过加速批准上市的新药。而且自从2014年以来,每年通过加速批准这条终南捷径上市的药物数量都保持在两位数(图1)。
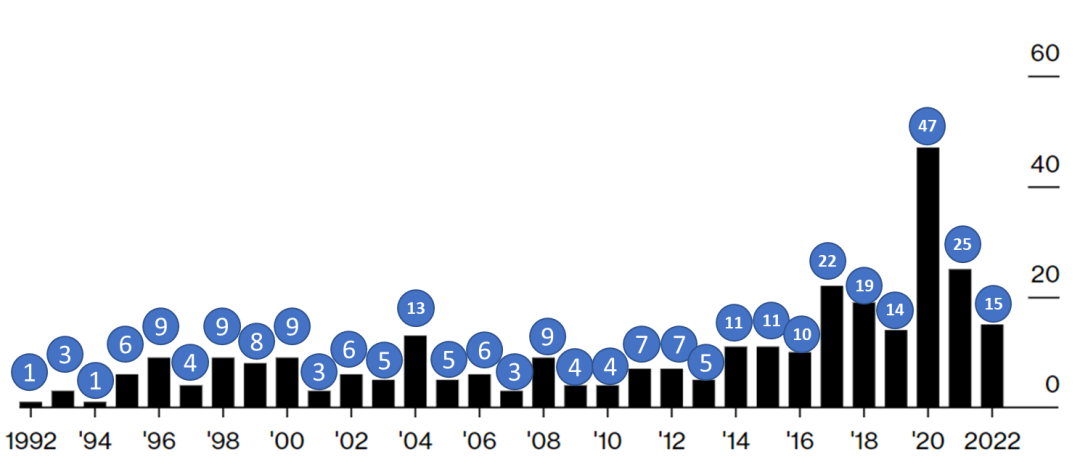
图1. FDA加速批准药物每年数量图。(数据来源:Bloomberg)注:同一款药物可以针对不同适应症获得多次加速批准。
Exondys在2016年批准时, FDA知道做出了争议决定。很多专家认为dystrophin蛋白质水平的小幅增加不足以帮助患者,但患儿家长们极力争取批准。尽管Sarepta也在欧洲寻求Exondys的批准,但EMA在2018年果断拒绝了它,并称相关研究数据“不能表明该药有效。” 虽然Sarepta对该决定提出上诉,但没有能够令EMA回心转意。Exondys上市之后,Sarepta又通过加速途径获得了FDA批准的两种治疗肌营养不良症的药物。
很多业内人士对于Exondys的上市给予了非常严厉的抨击,非营利性智库机构国家卫生研究中心(National Center for Health Research)主席Diana Zuckerman毫不留情地说:“Sarepta 是绝对离谱的,并且缺乏任何监督或执法形式的典型代表”。尽管Sarepta信誓旦旦地声称,它将“完全致力于”完成Exondys的批准后验证性试验,但对于延误的事实,它们还是起早贪黑地找出了多种理由,包括FDA的一些要求。除了Exondys之外,Sarepata的其他两种药物的验证性试验已经注册,预计将于 2025 年完成。
根据Sarepta的说法,Exondys的成功使该公司能够筹集到数十亿美元,并将其投入到基因治疗研究和制造中,以帮助更多的患者(即5月12日进行Adcomm审议的SRP-9001)。这似乎是“他山之石,可以攻玉”的理论。玉是不是玉先抛开不论,难道不要先把“疯狂的石头”的事情做个了结吗。
3 围绕加速批准的争议是什么?
关于加速批准有很多争议的话题,其中批准后研究是关注度最高的内容之一。Post-approval study试验经常被制药机构延迟。在某些情况下,它们甚至可能需要十多年才能完成这项研究。此外,一些获得加速批准的药物后来被证明对患者无效或不安全,但其药物已经在市面上流通多年。除了最明显的耽误患者病情之外,很多通过加速批准上市的药物价格奇高,例如被FDA加速批准的阿尔茨海默病药物Aduhelm(阿杜卡努单抗),上市后就立即引发了广泛的巨大争议。其制造商百健(Biogen)已于2022年4月从EMA撤回了其上市许可申请(MAA)。
EMA的人用药品委员会 (CHMP) 表示,所提供的数据不足以获得上市许可。但隔江犹唱后庭花的FDA至今尚无动作。批评者指出,Aduhelm价格如此昂贵,但包括政府、私人付款人和消费者在内的支付人,在没有明确临床益处的情况下还在为这些高昂的费用买单,落得人财两空的结局。加速审批最初是制药商向患者提供新药的一种相对“委婉晦涩”的方式,但现在已成为了一些公司的商业模式。批评者认为 FDA 没有采取足够的措施,来迫使制药公司为他们加速批准上市的药物提供确凿的数据。
4 祖宗之法必须变
加速批准改革箭在弦上
加速批准可以使得很多关键新药更快上市,恩泽患者,这个初衷并没有问题。但那些政策的受益者正在钻营的漏洞,必须尽快堵上。首当其冲的问题就是批准后研究的时限。FDA发言人称,加速批准是“为有严重和未满足医疗需求的患者提供有希望的治疗的重要途径”,但需要及时进行后续研究。一些政策提案认为,批准后研究应该设立期限,以防制药公司三心二意地磨洋工。并且如果制药公司没有及时进行验证性试验,或者如果试验结果表明安全性或有效性不佳,FDA需要明确的指导方针,规定FDA应该如何以及何时将药物拿下。
众议院和参议院的法案,都将要求FDA和药物制造商在相关药物加速批准之前,应该就批准后研究条件达成一致,并要求制造商定期地更新研究进展。该提案还将授予FDA权力,可以让FDA要求在药物获批之前就开始进行该类研究,并允许使用确凿的证据来支持这些研究。
如今的法律已经授予FDA权力,在申请者未按照due diligence(尽职调查)进行批准后研究,包括未验证临床益处,产品被证明不安全或无效,或者药物赞助商传播虚假或误导性宣传材料的情况下,FDA有权撤销药物的加速批准。然而,众议院和参议院的提议都试图简化并加快FDA撤回药物批准的程序。
图片来源:MEDPAGE TODAY
5 除恶务快
无效加速批准药物尽快下架
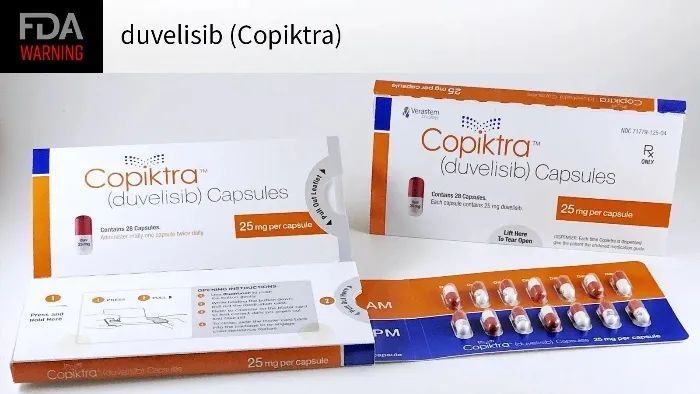
每天都有很多患者在服用加速批准上市的“疑似有效”的药物。耶鲁大学医师 Reshma Ramachandran 说,2020 年,她的一名患者在使用一种已获得加速批准的药物Copiktra(duvelisib)治疗滤泡性淋巴瘤后出现了结肠炎症。Ramachandran 说,患者需要手术切除部分结肠,但癌症并未治愈。2022年4月,该药物的制造商Secura Bio Inc.宣布撤回对该类型癌症的加速批准。Secura Bio Inc. 表示,撤回不是基于安全问题,而是它无力承担获得全面批准所需的更大规模试验的费用。Copiktra 目前仍在市场上销售,用于治疗其他类型的癌症。
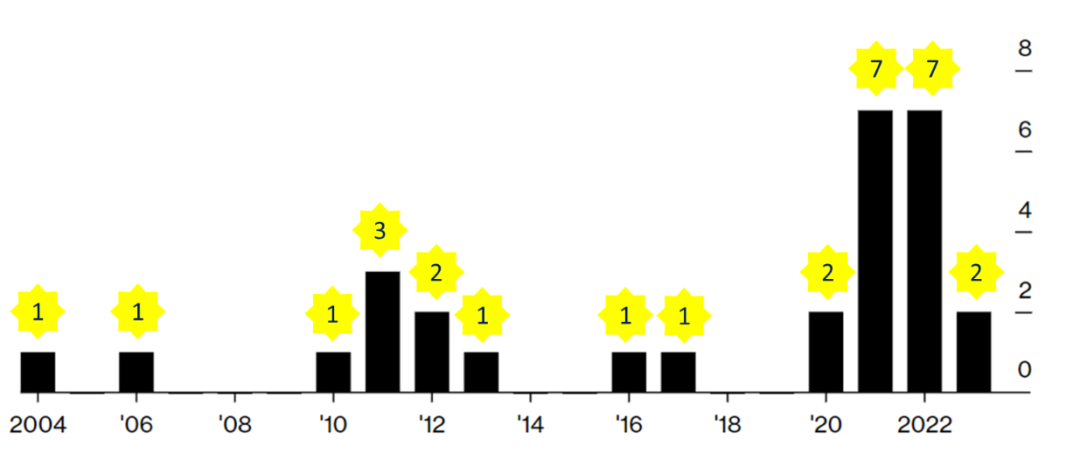
图2. 加速批准药物退市年份分布(数据来源:Bloomberg)。注:计数包括完全退出市场的药物和留在市场上用于其他用途的部分退出药物。具有多个批准的药物,在不同年份撤回的情况,每年计算一次。
随着加速批准引发的争议越来越大,FDA也在努力地自我纠错,让那些未能完成,或者未能通过验证研究的药物尽快退出市场。2022有7种药物退市,与2021年持平,为有史以来最多(图2)。下架是由公司自愿完成的,通常是应FDA的要求,但并非总是如此。
在过去的三十年里,大约有300款新药通过加速批准的途径问世,它们中间大部分用于抗癌药物。在某些年份,超过20%的上市新药都得益于这条高速公路。效仿美国,其他一些国家也制定了快速审批机制。但根据FDA官员的2022年审查,在日本和加拿大以外的大多数国家,这条批准捷径都设定到期时间。欧盟有一条加快的途径,但它一次只允许一种药物进入市场一年,要求公司更快地获取证明治疗有效的数据。
— 结语 —
允执厥中,加速批准的红与黑

图片来源:US.gleevec.com,新华网
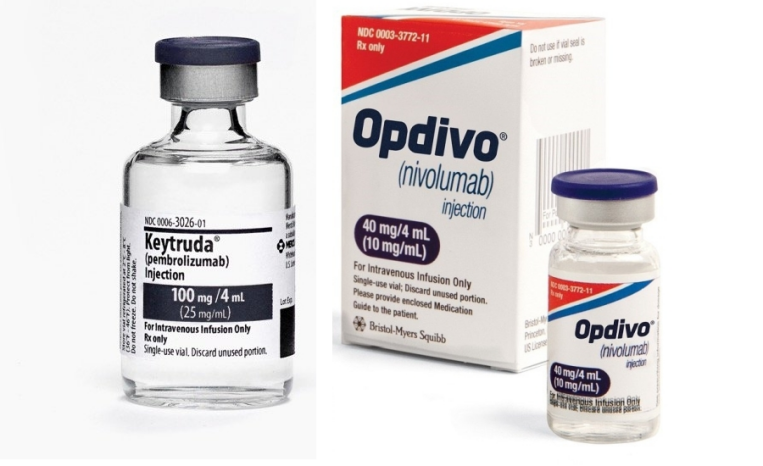
对于加速批准,人们应该用辩证的眼光看待。尽管存在很多争议,但是不能否认它的积极作用。《我不是药神》中的诺华白血病药物格列卫(Gleevec)就是2003年通过加速批准的途径上市的。加速批准也扩大了重磅炸弹癌症免疫疗法的使用,例如Merck & Co.的Keytruda和百时美施贵宝的竞争药物 Opdivo。但即使是这两款在多种类型的癌症中都非常得到有效并充分证明的药物,它们的针对某些癌症类型的加速批准后来也遭到撤回。
图片来源:FiercePharma
上文提到了Biogen的阿尔茨海默病药物Aduhelm 所引发的巨大争议,它的两项大型试验产生了相互矛盾的结果:一项研究表明它有效,而另一项则没有。FDA的Adcomm于2020年11月投票反对通过传统途径批准。但仅在7个月之后,FDA在患者权益倡导者的压力之下不可思议地通过加速批准的途径获准了Aduhelm的上市。FDA慷慨大方地授予Biogen八年的时间进行来进行第三次大型批准后研究,目前正在进行之中。

图3. 美国纳税者用在未经验证的加速批准药物上的费用。注:包括验证性批准后研究至少延期6个月的项目。百时美施贵宝表示,FDA 批准其Opdivo 试验延期。4 月 6 日,在研究未能证实 Makena 有效后,FDA 将 Makena 下架。(数据来源:美国卫生与公共服务部监察长办公室;图片来源:Bloomberg)
在Aduhelm引发的轩然大波之后,卫生与公众服务部监察长办公室调查了FDA的计划花费了纳税人多少钱。根据他们的报告,联邦Medicare以及Medicaid Program在2018年至2021年期间,在那些用于加速批准但验证试验被推迟的药物上,总共花费了超过180亿美元。
耶鲁大学的Ramachandran在谈到加速批准途径时说:“人们可能会在他们生命的最后一年里接受一些最终被证明是无用的东西。他们本可以尝试别的疗法。”所以,钱不是唯一的问题。
参考文献:
1.The Accelerated Approval Pathway for New Drug Therapies: Controversies and Proposed Fixes. The Commonwealth Fund. 14. 07. 2022.
2.Secura Bio, Inc.; Withdrawal of Approval of Relapsed or Refractory Follicular Lymphoma Indication for COPIKTRA. Federal Register. 13. 04. 2022.
3.Myshko, D. Biogen Withdraws European Application of Aduhelm. Formulary Watch. 22. 04. 2022.
4.Langreth, R. et al. Drug Companies Are Minting Billions on Unproven Treatments With FDA Shortcut. Bloomberg. 15. 05. 2023.
5.FDA Grants Accelerated Approval for Alzheimer’s Drug. FDA. 07. 06. 2021.
6.Aduhelm: Withdrawal of the marketing authorisation application. EMA. 22. 04. 2022.
 产业资讯
MedTrend医趋势 2025-05-12
80
产业资讯
MedTrend医趋势 2025-05-12
80
 产业资讯
医药时间 2025-05-12
82
产业资讯
医药时间 2025-05-12
82
 产业资讯
研发客 2025-05-12
81
产业资讯
研发客 2025-05-12
81
 微信公众号
微信公众号 热门资讯
热门资讯 热点标签
热点标签