

 产业资讯
产业资讯
 CPhi制药在线
CPhi制药在线  2022-04-08
2022-04-08
 7051
7051

2022年03月31日,国家药监局官网发布再次公开征求《药品上市许可持有人检查要点(征求意见稿)》意见,征求意见截止日期2022年4月30日。这是国内专门针对药品上市许可持有人检查指南,这是国家药监局为贯彻实施《中华人民共和国药品管理法》《中华人民共和国疫苗管理法》,全面落实药品上市许可持有人对药品研制、生产、经营、使用全过程中药品的安全性、有效性和质量可控性的主体责任,进一步规范对药品上市许可持有人的监督检查工作。该检查指南弥补了我国对MAH现场检查在法规层面和技术层面的空缺,进一步完善MAH监管长效机制。本文对《药品上市许可持有人检查要点(征求意见稿)》中主要内容进行了分析,建议药品上市许可持有人开始对照检查吧,如有补充欢迎文末留言分享。
一、MAH制度历史沿革及相关法规概述
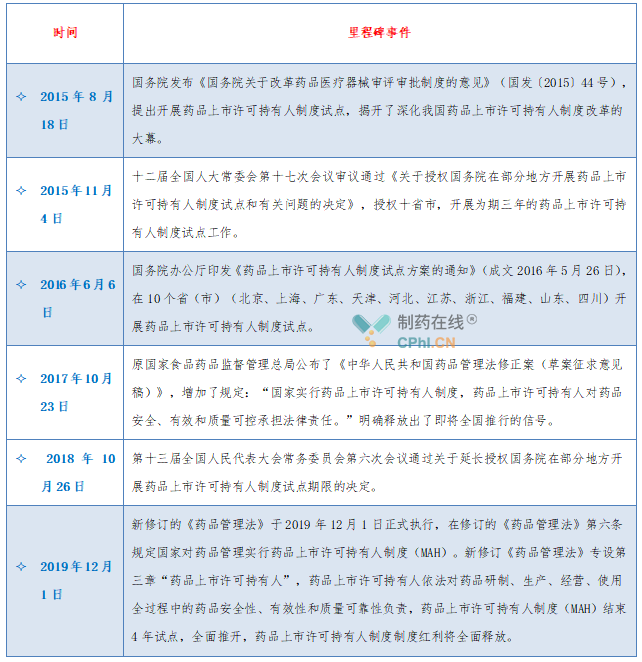
我国药品上市许可持有人制度是一个从无到有、不断完善、循序渐进的过程,新版《药品管理法》实施后,标志着药品上市许可持有人制度全面落地实施,各阶段政策法规摘录见下表:
二、MAH制度文件化
持有人应当建立覆盖药品研制、生产、销售、使用全过程的质量保证体系,持续强化的质量控制和质量保证能力,依法对药品研制、生产、销售、使用全过程的安全性、有效性、质量可控性负责,人员是关键,硬件是基础,软件是保证。从目前的实际情况来看,我国许多MAH在硬件建设方面普遍投入充足,人员素质也相对比较高,但软件系统的完善是当前进行质量保证体系的首要任务。《药品上市许可持有人检查要点(征求意见稿)》对于文件体系有了更具体的要求:持有人应当建立保证药品全生命周期主体责任的规章制度。委托其他企业进行药品生产、销售相关活动(包括药品储存、运输)的,相关制度应当与受托企业的质量管理体系文件有效衔接,并按照规定形成相关记录或报告。包括但不限于:
(1)药品生产场地管理文件;
(2)委托协议和质量协议;
(3)持有人对受托企业的审核程序、现场审核报告及记录;
(4)研制、生产、销售监督管理程序与记录。
(5)药品质量回顾分析制度及药品质量回顾分析报告;
(6)药品生产工艺规程、空白批生产记录、质量标准及检测程序;
(7)受托生产企业共线生产药品列表及风险评估报告(或关于避免污染及交叉污染的相关程序与记录/报告);
(8)药品偏差、变更控制、自检、不合格品处理、纠正与预防措施、质量投诉、退货、召回、物料与产品、确认与验证、稳定性试验、数据可靠性、培训、员工健康、上市放行、追溯等管理程序及记录;
(9)生产关键物料合格供应商名单;
(10)质量信息沟通及处置的规定与沟通记录;
(11)药品安全事件处置方案与培训、演练记录;
(12)药物警戒管理程序与记录;
(13)药品年度报告管理程序与记录;
(14)药品上市后风险管理计划与记录;
(15)短缺药品停产报告管理程序与记录(针对短缺药品的持有人)。
建立一套文件系统成功与否,关键是文件总目录的确定,先拟定文件目录草案,这样可以进行文件编制分工,明确目标、掌握进度,在编制过程中可以结合公司实际情况对文件目录进行修改,文件目录至少涵盖岗位职责类文件、技术标准类文件、管理标准类文件、操作标准类文件、记录类文件等文件目录。MAH文件体系包括的制度清单建议如下:

参考文献
[1]www.nmpa.gov.cn
 产业资讯
瞪羚社 2025-05-01
48
产业资讯
瞪羚社 2025-05-01
48
 产业资讯
医药经济报 2025-05-01
49
产业资讯
医药经济报 2025-05-01
49
 产业资讯
药渡Daily 2025-05-01
47
产业资讯
药渡Daily 2025-05-01
47
 微信公众号
微信公众号 热门资讯
热门资讯 热点标签
热点标签