

 产业资讯
产业资讯
 制药视界
制药视界  2024-03-07
2024-03-07
 5084
5084
脊髓性肌萎缩症(SMA)临床表现为进行性、对称性,肢体近端为主的广泛性弛缓性麻痹与肌萎缩,智力发育及感觉均正常。两种经FDA批准的SMA治疗方法(nusinersen和risdiplam)是通过修饰SMN2的剪接来增加SMN蛋白的产量,需要连续给药。
2022年6月17日,诺华在Nature Medicine上发布了一篇文章,详细描述了Onasemonogene abeparvovec(商品名Zolgensma)的III期SPR1NT(NCT03505099)试验。这一药物最初由AveXis研发(后来该公司被诺华收购),是一种基因替代疗法,于2019年5月24日获得美国FDA批准上市。该药物通过腺相关病毒9型(AAV9)载体将人的SMN cDNA递送到靶运动神经元细胞中,使其得以表达SMN蛋白。

SPR1NT研究:SMA病因与进展
脊髓性肌肉萎缩症(SMA)是一种常染色体隐性神经肌肉疾病,由SMN1(存活运动神经元1)基因的双等位基因缺失或致病性变体导致的存活运动神经元(SMN)蛋白缺乏引起。SMN2是SMN1的同源基因,产生少量SMN蛋白部分补偿SMN1的缺失。SMN2的拷贝数与发病和严重程度相关,有三份SMN2拷贝的患者,发育为7个月至18个月发病的中度SMA 2型的可能性高达54%,为1型或症状较轻的3型的可能性分别为15%和31%。
患有SMA 2型的未经治疗儿童在13岁之前会经历相对快速的神经肌肉衰退,在成年后逐渐衰弱。患者能够独立坐着,但很难站立,也不能独立行走。随着年龄的增长,几乎所有2型SMA患者都会出现吞咽困难、关节挛缩、脊柱侧凸和限制性肺病,一些患者可能会失去独立坐着的能力。3型SMA的致残程度低于2型。
因此,在SPR1NT的29名参与者中,包括14名有两份SMN2的儿童和15名有三份SMN2的儿童,所有受试婴儿的神经肌肉功能正常,能够正常吞咽和呼吸,尚未出现SMA临床症状。本篇文章重点关注15名携带三份SMN2的SPR1NT参与者,并提供有关该人群中新生儿AAV9载体输注的重要新疗效和安全性数据。
SPR1NT是onasemnogene abeparvovec治疗有SMA 1、2或3型风险的症状前期婴儿的第一项单臂III期研究,受试婴儿以单次静脉输注的形式接受给药。这项试验侧重于疗效指标,如运动里程碑(motor milestone),通过与正常发育基准相比较来衡量病患在没有机械干预的情况下生存和茁壮成长的能力,同时还将疗效和探索性措施与儿童神经肌肉临床研究(PNCR)自然史人群进行了比较。
结果
主要和次要运动能力
所有15名儿童在24个月大之前的就诊中,都做到了独立站立。在24个月的研究访问中,所有儿童都保持着独自站立的能力。有14名儿童在24个月内某一次就诊中至少独立行走了五步。相比之下,PNCR 81例SMA患者中只有19例做到了独自站立,17名可以独立行走。
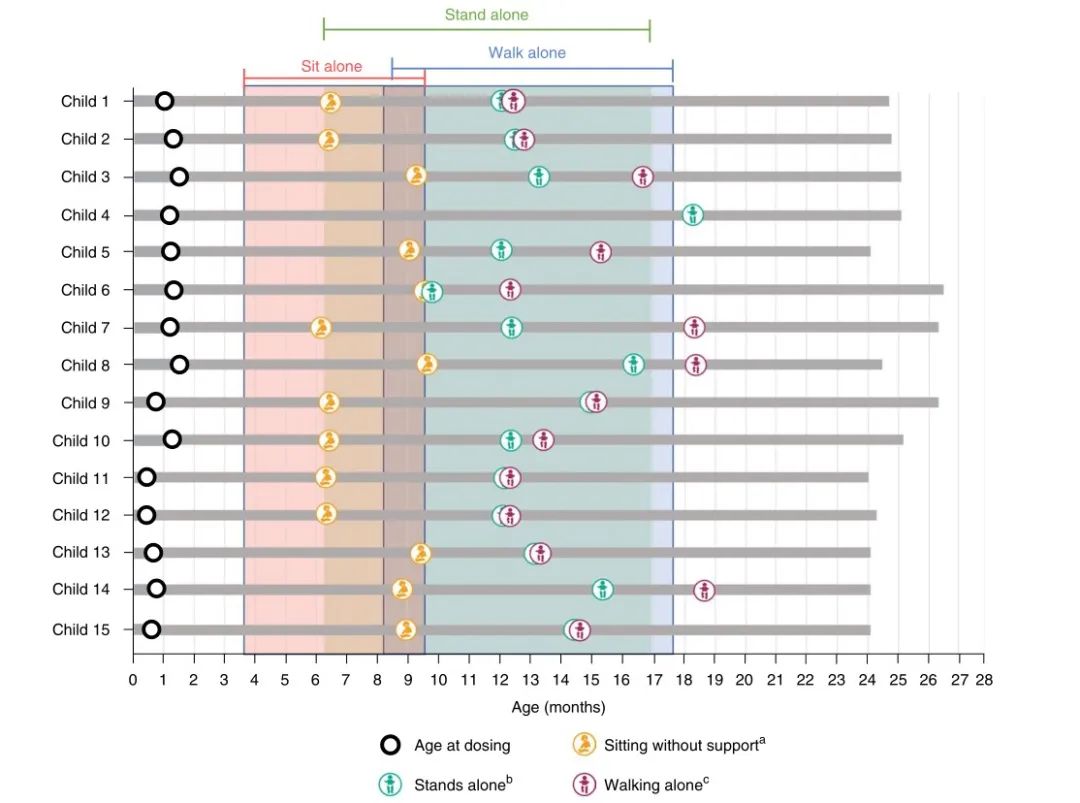
▲受试儿童的发展运动情况 图片来源:参考资料1
运动状况精细分析
与年龄匹配的参考人群相比,Bayley婴幼儿发展量表(BSID)提供了更精确的发展评估,总体和精细运动原始分数的增量增长通常参考正常人群。使用BSID对所有15名儿童进行充分评估(有一名儿童未完成),将原始分数转换为标准平均值为10、标准偏差(s.d.)为3的量表分数,这样,4–16的量表分数就可以捕捉到正常运动发育的3%到97%。三拷贝队列中的所有15名儿童的量表得分均为≥4。大多数被评估的儿童在就诊时都符合标准。在24个月的随访中,所有10名接受评估的儿童的量表得分均为≥4。
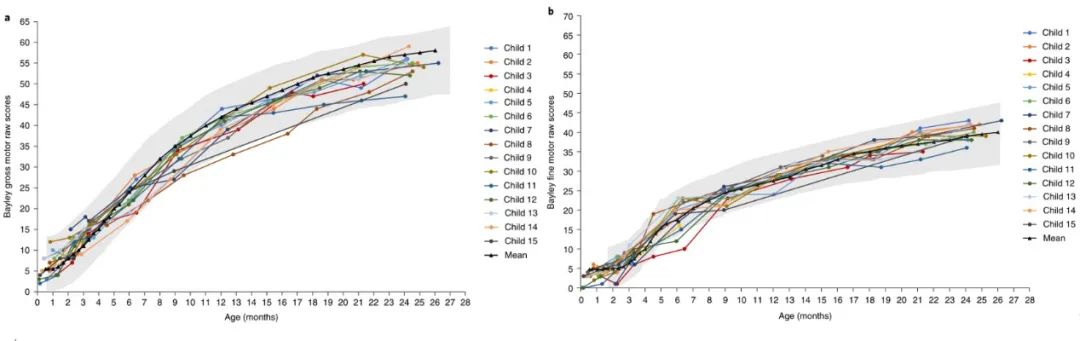
▲受试儿童生长发育表 图片来源:参考资料1
生长发育状况
所有15名儿童在14个月大时均存活,在整个试验期间,没有儿童需要任何类型的机械呼吸支持(例如,咳嗽辅助、双水平气道正压或有创通气支持)。与此同时,15名儿童中有10名体重处于或高于世卫组织标准的前百分之三,并且在研究结束时,所有儿童都处于或高于该百分位。在研究期间都没有儿童需要喂食管来进行辅助喂食。
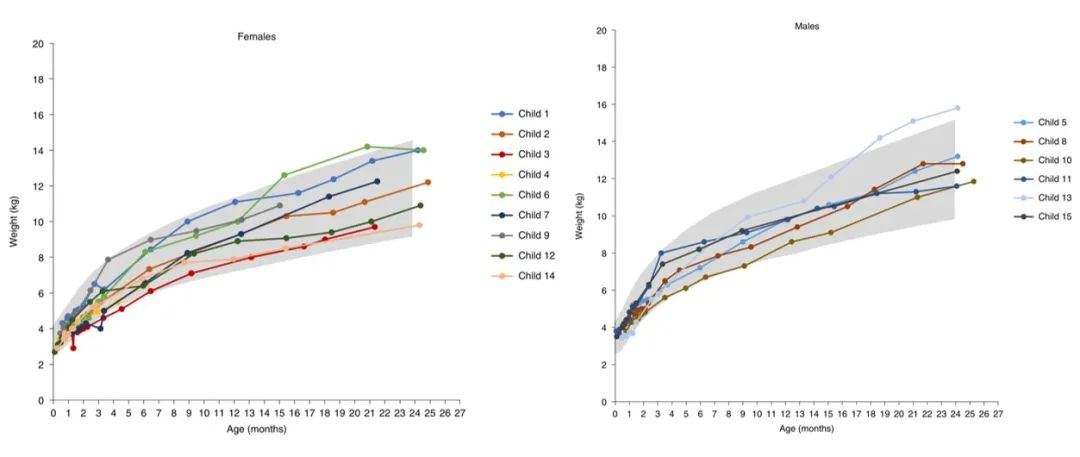
▲Bayley量表原始分数 图片来源:参考资料1
安全性
研究人员通过监测不良事件、体检、肺部检查、生命体征、体重和身高测量、12导联心电图、24小时动态心电图监测、超声心动图、吞咽试验、实验室评估和输液部位的照片来评估安全性。
在试验过程中共报告了166例治疗突发不良事件(TEAE)。每个孩子至少经历过一次TEAE,其中三个儿童的TEAE报告为严重。研究人员认为15名儿童中有8名经历的TEAE与研究治疗有关,但均不严重并且得到了及时有效的处理。
结论
SPR1NT证明,单次静脉注射onasemnogene abeparvovec可促进SMA症状前新生儿的运动发育,这些症状前新生儿具有SMN1双等位基因缺失和三份SMN2拷贝,主要存在SMA 2型风险。如果不进行治疗,这些儿童中的大多数都不会取得比独立保持坐姿更大的运动里程碑,无法独自站立和独自行走。接受了Onasemonogene abeparvovec治疗的儿童显示出与健康儿童相同的运动发育模式。
此外,SPR1NT中没有儿童需要机械喂养或呼吸支持,这表明症状前基因治疗有可能预防典型SMA 2型的某些肌肉骨骼、肺部和生长并发症。这代表着在疾病早期,可以实现向更温和的SMA表型,甚至向正常运动发育的深刻转变。
研究人员还观察到,静脉注射Onasemonogene abeparvovec治疗症状前新生儿表现出良好的安全性。
总结
目前,诺华Zolgensma已在国内启动临床试验,是全球III期临床STEER研究的中国部分。
二十年前,人类基因组计划承诺了新的诊断和治疗方法,其基础是确定疾病的潜在遗传机制。最终,基因组学研究旨在改变医疗实践,从被动的立场,即呈现疾病的迹象和症状,及时治疗,转变为主动的立场,深入了解基因组中潜在的脆弱性,使提供者能够预测未来的健康风险,并应用精确的干预措施,保持人们的健康。目标是在正确的时间为正确的患者找到正确的治疗方法,从而预防疾病和残疾。随着SMA分子基础的发现、有效治疗和最佳干预时机的确定,SMA儿童的这一目标可能很快就会实现。
参考资料:
1.Strauss, K.A., Farrar, M.A., Muntoni, F. et al. Onasemnogene abeparvovec for presymptomatic infants with three copies of SMN2 at risk for spinal muscular atrophy: the Phase III SPR1NT trial. Nat Med (2022). https://doi.org/10.1038/s41591-022-01867-3
 产业资讯
药通社 2026-06-06
460
产业资讯
药通社 2026-06-06
460
 产业资讯
赛柏蓝 2026-06-06
360
产业资讯
赛柏蓝 2026-06-06
360
 产业资讯
摩熵医药 2026-06-06
378
产业资讯
摩熵医药 2026-06-06
378
 热门资讯
热门资讯 微信公众号
微信公众号