

 产业资讯
产业资讯
 同写意
同写意  2024-08-16
2024-08-16
 1138
1138
26岁的Saritee Sanodiya,无数天都在怀疑自己能否过上“正常”生活。成长过程中,她经常因为血红蛋白水平突然下降等问题而去医院。高烧、剧烈头痛、极度疲劳、呼吸困难,这些症状可以持续几天甚至数周。“它们突然出现,这种痛苦很难描述,无法忍受。
有时你会失去活下去的想法。”医生通过输血治疗贫血,使用止痛药来缓解病人身体的不适。症状会逐渐消退,但几个月后又卷土重来。直到2018年,Sanodiya在第一次孕检时,进行了镰状细胞病(SCD)血液检测,笼罩在她身上的疑云才消散。阳性结果意味着,基因突变让她携带了非正常的红细胞。
Sanodiya从未听说过这种疾病。其实,跟她拥有相似遭遇的人恒河沙数,尤其在印度——这里是世界上SCD发病率最高的国家之一。好消息是,2023年FDA批准了两款基因疗法,能从根本上解决这种疾病。
不过,一针高达上百万美元的售价,也直接宣告用得上它们的人群有限。许多中低收入国家的卫生保健系统,以及像Sanodiya这样的患者,都无法负担这些成本。目前,印度的研究人员试图开发类似的产品,并以更低的价格向国内市场提供。低价版基因疗法并不是印度在CGT领域的首次尝试。
3月,Nature的一篇文章就介绍,当地一家药企ImmunoACT开发出的CAR-T疗法,单次治疗费用仅3-4万美元,几乎只有美国同类产品的十分之一。一系列因素促成了上述结果,例如,印度有特殊的专利保护法律、仿制药开发经验。可这种成功会不会在SCD基因疗法上得到复制,仍是一个未知数。
性命攸关
The Lancet Haematology介绍,作为一个多世纪以前科学文献中记录的首种单基因疾病,SCD仍然是一种发病率和死亡率很高的疾病。
根据相关量化研究,科学家估计,全球每年有超过30万名婴儿在出生时患有纯合子型SCD,预计到2050年,患者规模将增加到40万以上。
正常情况下,人的红细胞就像一个双凹圆盘,形态可变。但基因突变后,红细胞呈镰刀状,僵硬而粘稠,容易堵塞血管。与寿命约为120天的正常红细胞不同,这些镰状细胞只能存活10-20天。
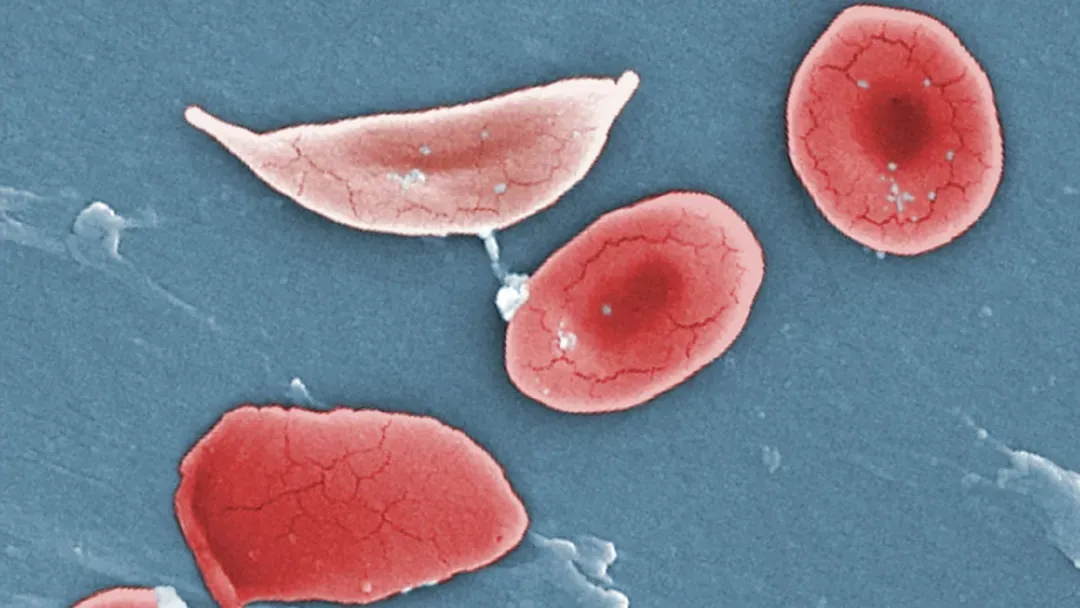
彩色显微镜图像显示镰状细胞(上)和镰状细胞性贫血患者的正常红细胞
当血液循环中的血细胞减少,就会出现贫血。同时,肝脏的血液过滤能力难以应付大量死亡的镰状细胞,分解副产物——胆红素——便开始积聚,从而又导致黄疸。所以,这些非正常的红细胞让人产生衰弱性疼痛,并对大脑、肝脏、肾脏、心脏、肺和关节等器官造成渐进性损害。Sanodiya确诊以来,除了怀孕那几个月,她每天都在服用羟基脲。
这是一种应对SCD最常用的药片,有助于保持红细胞形状和柔韧性,减少血液中镰状细胞数量。理论上,每日使用羟基脲可以减少疼痛发作,并最大限度地减少输血和医院就诊的需要。不过,正如波士顿儿童医院和哈佛医学院的Vijay Sankaran指出的,“并不是每个患者都有相同的反应”。例如,Sanodiya就一直饱受折磨。今年4月,她的眼睛明显发黄,一叠的医疗记录显示,因为SCD不知跑了多少次医院,做了数不尽的胸部X光和血液检查。
尽管医生们谨慎地尝试增加Sanodiya的羟基脲剂量,对于患有严重SCD的人,骨髓移植才是一种有效的治愈方法——在FDA批准两款基因疗法之前,这某种程度是唯一的希望。该手术涉及用匹配捐赠者(通常是兄弟姐妹)的健康细胞替换患者骨髓中的异常干细胞。
印度赖布尔巴尔科医疗中心血液学家Dibyendu De表示,想要找到匹配的捐赠者非常困难。此外,如果没有经济援助或政府补贴,大多数人都无法负担手术治疗费用。Sanodiya面临的情况正是如此。因为在私立医院接受紧急护理,她的家庭眼下负债累累。欧美市场的基因疗法成本高昂,客观上,却给印度的SCD患者提供另一种看得到的可能。
“我们距离目标还很远,但事情正在朝着正确的方向发展。”Debojyoti Chakraborty是德里基因组学和综合生物学研究所的分子生物学家,正领导着一个开发SCD基因疗法的团队。他相信,这个目标在印度也可以实现,毕竟至少从机制上,海外同行已成功走了一遍。
基因疗法
多年来,研究人员一直在寻找治疗SCD和β-地中海贫血——另一种遗传性血液疾病——的更优方案,以提高胎儿血红蛋白(HbF)水平。
HbF在出生时很高,并随着儿童长大而降低,被成人血红蛋白在很大程度上取代。不过,SCD患者的骨髓通常会产生一种异常形式的血红蛋白。增加HbF的数量,有助于阻止这种镰状血红蛋白的聚合,从而减轻疾病症状并延长寿命。
2008年,Sankaran及其同事取得了突破。他们发现,沉默一种名为BCL11A的基因,可以刺激成体细胞产生HbF。“这是一个重要的里程碑。”Sankaran评价说。
为了安全地靶向BCL11A治疗SCD,该团队确定了基因中对BCL11A表达至关重要的区域,特别是在红细胞中。他们使用CRISPR基因编辑工具在这个区域进行切割并破坏DNA序列,降低BCL11A的表达,增强HbF的产生。
波士顿儿童医院和哈佛医学院的医生兼干细胞生物学家Stuart Orkin表示,这是一种迂回的方法,但它确实带来了首批上市的SCD基因疗法。
这种疗法名为Casgevy,由CRISPR Therapeutics和Vertex Pharmaceuticals开发。它需要从骨髓中取出造血干细胞,在BCL11A基因的精确位置切割两条DNA链,然后将这些编辑过的细胞输入患者体内。
数据显示,Casgevy能将成人红细胞中的HbF水平提高到约30-40%。临床试验接受治疗的30人中,有29人在至少12个月内没有出现严重症状,并且没有人需要住院治疗。
患者的长期状况如何还有待观察。但Sankaran说,这种疗法并不温和,患者必须接受彻底的化疗以清除原生干细胞,为编辑过的干细胞腾出空间。该过程可能导致不孕,患者患血癌的风险不大却会增加。
上述担忧,也适用于另一种获批的SCD基因疗法,Lyfgenia。它由Bluebird Bio开发,将产生健康血红蛋白的正确基因传递给患者的造血干细胞。这个过程在实验室中进行,然后将经过修改的细胞送回患者体内。
临床结果显示,在接受Lyfgenia治疗的33名SCD患者中,有32名在治疗后6至18个月内没有经历严重不良事件。
从2023年到2024年,中东的巴林、沙特阿拉伯,以及美国、英国、欧盟等地的监管机构批准了至少一种此类疗法,用于治疗12岁及以上患有严重SCD的患者。但它们价格昂贵,生产制造的时间长。治疗过程可能长达一年。
“到2025年,如果我们能在美国使用基因疗法治愈20-50名SCD患者,那就算幸运了。”Akshay Sharma表示。他是圣犹达儿童研究医院的儿科血液学家,负责过包括Casgevy在内的SCD治疗临床试验。
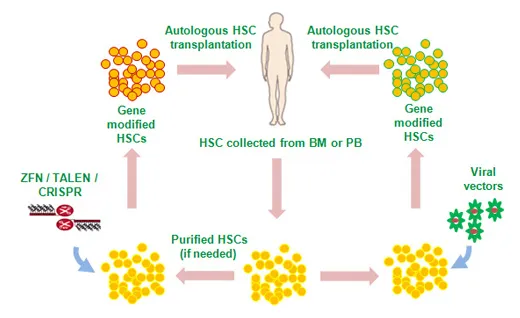
针对血红蛋白疾病的基因疗法
目前获批的基因疗法,似乎也不太可能惠及中低收入国家的患者,虽然后者才是这种疾病的主要人群。
“这些公司对在非洲、印度或巴西进行试验不感兴趣,”Sharma解释道,“它们无法在那里收取200万至300万美元的费用;它们无法从中获利,所以不想这么做。”
将基因疗法引入这些地区,必须由这些地区的科学家自己来完成。
背后不仅仅是成本问题。“如果我们不从头了解它们(基因疗法)如何与不同人群的细胞生物学和遗传学相互作用,我们就会浪费时间,以为我们开发出了一种万能药。”第三方组织全球基因治疗倡议的联合创始人Jennifer Adair说。
不再忽视
SCD所涉及的突变,原本是为了帮助疟疾高发地区的人类抵御感染。科学家已证明,镰状细胞不利于导致疟疾的寄生虫生长。但同时,它也引起SCD的肆虐。
世界上超过一半的SCD患者生活在三个国家——尼日利亚、刚果民主共和国、印度。与世界上许多其他地区一样,几十年来,这种疾病在印度一直被忽视。
印度SCD专家Dipty Jain表示,医护人员缺乏意识,意味着患者可能多年得不到诊断或被误诊,难以衡量这种疾病的真正负担。一些估计表明,可能有超过100万印度人受到影响,很大一部分来自部落社区,以及其他历史上处于弱势地位的群体。
造成这种现象的原因,也许是在某些群体居住的地区疟疾肆虐,继而携带突变基因。此外,印度有着同族或同种姓通婚的古老习俗,为突变携带者生下患有SCD的孩子创造了更大概率。
不过,Jain表示,SCD并不只发生在部落或其他边缘群体。伦敦帝国理工学院研究SCD健康负担的流行病学家Frédéric Piel称,全球卫生界一直对印度SCD有误解,认为它的主要症状较轻。但实际上,印度中部许多患者都出现了严重症状。印度科学家花了很长时间,才改变了这种看法。
需要指出,在实际情况中,一些医生对这种疾病仍然缺乏足够重视。
印度非营利性镰状细胞组织全国联盟秘书Gautam Dongre介绍,SCD患者经常因反复发作的疼痛而到医疗中心就诊,医生和护士可能会尽量减轻他们的痛苦,而不是优先为他们提供紧急护理。
让这种疾病更加不为人知的是,它迫使人们退出社会。对于Jitendra Kumar Sahu,尽管他想学习,但辍学是他唯一的选择。
因为SCD,Sahu每年几乎只能上学几个月。他很少参加体育或其他需要体力的课外活动,而且这种疾病对他的髋关节损害非常严重,以至于这个21岁的年轻人,在今年4月进行了髋关节置换手术。
“我感到很难过。”Sahu说,由于他可能无法工作,常常看不到未来的希望。他也担心家人正遭受的经济困难,医疗费用让他们负债累累。
SCD还带有社会耻辱感。许多年轻人的父母,尤其是女性会担心,患有SCD的孩子可能很难结婚。这种恐惧可能导致一些家庭不愿透露病情,或避免谈论患有这种症状的生活会是怎样。
其他情况下,耻辱感来自于公众的误解,即认为SCD只发生在社会弱势群体中。Dongre形容,如果不是这个群体的人被诊断出患有SCD,“他们会感到尴尬、害怕,要么不承认,要么不寻求治疗”。
精神健康负担是一个持续的挑战。应对终身疾病的挣扎、缺乏社会和医疗环境的支持以及经济困境,都可能导致人们感到沮丧、焦虑和抑郁。Dongre说,他的两位SCD患者同事最近自杀身亡。
2023年7月,印度政府启动了一项计划,旨在遏制SCD向下一代传播,并在2047年前基本消除该病的流行。计划的目标之一是提高社会的认识,并在2026年前对SCD流行的国内17个州多达7000万人进行筛查。
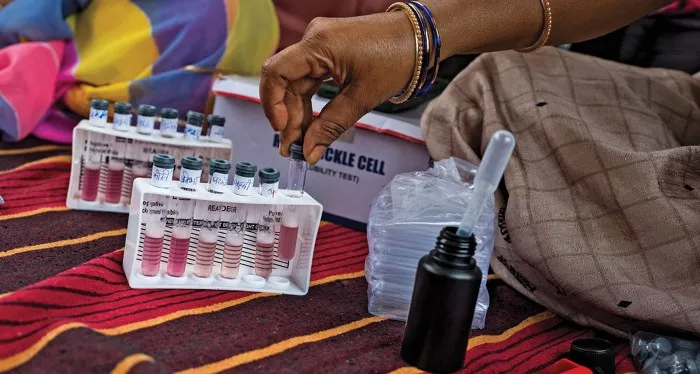
在溶解度测试中,镰状细胞的存在会使制备溶液变得浑浊
印度有超过100万的社区卫生工作者,全部由女性组成,这个团体被称为ASHA(印地语,意为“希望”)。她们是传播SCD意识和实现筛查目标的关键,尤其在印度农村地区。
Jain担任了国家镰状细胞消除任务的负责人,他表示,婚前或产前筛查将是关键。
印度政府希望提供随时可用的遗传咨询服务,帮助新婚夫妇或父母了解他们基因的兼容性,以及遗传SCD的几率。另一个目标,是确保患者能够享受到优质的护理,包括羟基脲和输血等。虽然它们并不治本,但对许多人来说,获得这些疗法并不容易。
作为计划的一部分,政府将在17个州内确定并升级一些三级医疗中心,以便为符合条件的患者提供骨髓移植服务。有的官员将视线转移到更有效的解决方案上:在印度,开发SCD基因疗法。
针对突变
今年4月,25名SCD患者与家人聚集在印度杜尔格的一家医院。与他们会面的,除了Chakraborty之外,还有同样来自德里基因组学和综合生物学研究所的化学家Souvik Maiti。
Chakraborty和Maiti是印度CRISPR基因编辑疗法项目的共同负责人,如果一切顺利,他们计划在2025年开始一项人体临床试验,并从杜尔格所在的恰蒂斯加尔邦招募3到5人参与I期临床。
不同于旨在提高HbF水平的Casgevy,也不像诱导产生具有健康血红蛋白的红细胞的Lyfgenia那样,印度团队的目标疗法,是直接纠正导致SCD的突变。
由于这是单一突变,“如果你纠正了它,你就会把SCD患者变成携带者”。换言之,接受治疗后的患者,将不用忍受SCD的各种严重症状。
对于CRISPR编辑工具,印度团队选择了一种经过改造的蛋白酶FnCas9,而不是业界更常用的SpCas9。
在2019年的一项研究中,研究人员报告称,FnCas9能以极高的特异性与需要编辑的DNA区域结合。但未经改造的FnCas9效率不佳,因此该团队进行加工,以保留其特异性并提高效率。
作为概念验证,他们在实验室中借助这种工程化蛋白酶,纠正了人类细胞中一种罕见遗传性眼病的突变。研究人员还使用基于FnCas9的工程化疗法纠正小鼠SCD的突变。不过,这些结果尚公开未发表。
该团队已在美国获得FnCas9的专利。Chakraborty表示:“专利让我们可以对外授权CRISPR系统。”他认为,这可能会激励印度Biotech有朝一日将相关疗法推向市场。
这种印度本土化改造的基因疗法售价拟定在6万美元左右,一个比现有的竞品要低得多的数字。但不得不说,许多患者仍然负担不起。Chakraborty寄希望于政府资助,以覆盖大部分剩下的费用。
与其他基因疗法类似之处是,临床医生需要首先将干细胞从患者的骨髓提取到血液中,然后收集这些细胞给到研究人员进行编辑,同时患者接受化疗以杀死剩余的骨髓干细胞。
在杜尔格举行的见面会上,Chakraborty向患者介绍了接受Casgevy或Lyfgenia治疗后的获益,以及可能由于化疗引起的生育问题。这些基因疗法是新疗法,因此很难知道患者是否持续感觉良好,以及是否完全治愈。

Chakraborty向患者介绍SCD基因疗法Chakraborty提到Victoria Gray,她是2018年接受SCD基因疗法的第一位患者,该疗法正是2023年获批上市的Casgevy。5年多以来,Gray都没有出现严重的疼痛症状,也无需住院。“我没有输过血,没有出现过危急情况,也不再依赖药罐。”她回顾说。
Chakraborty和Maiti领导的团队,并不是印度唯一一个开发SCD基因疗法的团队。韦洛尔基督教医学院的科学家一直在与埃默里大学合作,测试类似于Lyfgenia的方法,通过递送正确的基因来表达健康的成人血红蛋白或HbF。这些疗法似乎可以缓解小鼠的疾病症状。此外,印度研究人员还在开发另一种基于CRISPR的基因编辑技术。埃默里大学医学院细胞和基因治疗项目主任Trent Spencer表示:“目前我们正在与印度的一些公司洽谈,还有一些公司对实际生产基因治疗产品感兴趣,但目前尚未达成最终合作。”
早前的2月,德国公司Miltenyi Biotec也宣布,将在印度设立一个细胞和基因治疗中心。这家Biotech希望通过提供实践培训、专业知识和制造技术,来帮助印度的研究人员。它还与印度转化卫生科技研究所合作开发基因疗法,重点关注癌症和SCD。
打下价格
在美国,科学家正开展临床试验,以测试Casgevy和Lyfgenia对12岁以下儿童是否同样有效。这些研究尤为重要,因为SCD是全球5岁以下儿童死亡的第12大原因。
世界各地许多专注于基因疗法的科学家,也在尝试寻找比BCL11A更好的靶点来提高HbF水平。跟Chakraborty、Maiti所做的类似,他们也在努力纠正导致SCD的潜在突变。为了使治疗更安全,研究人员尝试绕过化疗的方法。不过,任何基因疗法都很难扩大规模。
Orkin认为:“如果我们的目标是减轻疾病负担,那就真的需要开发体内基因疗法,或者是一种更好的药丸。”
体内基因治疗指的是,将编辑后的基因直接递送到指定的细胞,而不必经过取出细胞、编辑再重新放回体内的过程。
在最近的一项概念验证研究中,费城儿童医院和宾夕法尼亚大学的研究人员使用脂质纳米颗粒——嵌有能识别特定细胞受体的抗体——将基因编辑工具送到小鼠的造血干细胞里。他们发现,这种“一劳永逸”的方法不需要化疗或干细胞移植,大大增加了健康血红蛋白的产生,而且血液中几乎没有形成新的镰状细胞。
“结果非常令人兴奋,不过仍处于初步阶段。”Thompson表示,这可能会使更多的患者可以使用这些疗法。
当然,体内基因疗法也是极具挑战性的。现阶段,Spencer警告说:“当你从人体中取出细胞,改变基因就已经够难的了;而当细胞还在人体中,选择性地改造细胞仍然非常非常复杂。”
另一方面,Sankaran认为,过分关注SCD的基因疗法,可能会影响到开发比羟基脲更好的口服药物。小分子药物比基因疗法更具成本效益,也更容易获得。
自2017年以来,FDA已批准了一种名为crizanlizumab的单抗,以及voxelotor、L-glutamine两种新的口服药物,用于治疗SCD。但就像羟基脲一样,它们无法治愈SCD,也不是全球都能轻易获得的。
“我们仍然认为,羟基脲可能是我们拥有的最有效的药物。”Sankaran说。
同时,对于印度许多SCD患者,Chakraborty、Maiti等研究人员所做的工作带来了一线希望。“我们的科学家正在努力开发一种治疗方法,这对我这样的患者和后代来说是一件大事。”Sanodiya期待说。
参考资料:
1.Sickle cell disease in India: The quest for a cure;C&EN's
2.镰状细胞病:易被忽视的全球健康问题急需采取行动;柳叶刀TheLancet
 产业资讯
深蓝观 2026-03-31
29
产业资讯
深蓝观 2026-03-31
29
 产业资讯
贝壳社 2026-03-31
28
产业资讯
贝壳社 2026-03-31
28
 产业资讯
同写意 2026-03-31
28
产业资讯
同写意 2026-03-31
28
 微信公众号
微信公众号 热门资讯
热门资讯 热点标签
热点标签