

 产业资讯
产业资讯
 中国医药创新促进会
中国医药创新促进会  2025-05-09
2025-05-09
 1918
1918
引言
当全球生物医药产业驶入深水区,创新药“出海”已不仅是企业扩张的必选项,更是中国医药产业升级的战略支点。日本作为全球第三大医药市场,与中国药企在靶点开发、成本控制方面优势互补,成为中国创新药企出海的重要一站。
中国医药创新促进会始终致力于深化中日医药产业合作,并在医药创新领域取得了一系列实质性成果。为全面助力中国药监、生物技术公司、投资界深入了解日本药品监管法规、申报流程、沟通交流以及生产和检查等相关事宜,我会联合研发客、上海市生物医药科技产业促进中心以及泰格医药,共同开设“出海日本”专栏,特邀日本法规监管领域的资深专家发布专业性文章。撰稿人包括著名的药品开发及监管专家植村昭夫博士、东内祥浩先生和高野哲臣先生。同时研发客主编毛冬蕾女士还将对日本政府、学术界以及中日两国业内专家进行访谈,共同探讨开发及监管热门话题。
中国药促会中日医药合作交流
联系人:马明尧
电话:13520846026
邮箱:mamy@phirda.com撰文| 医药研发达人主编 高野哲臣(t2T Healthcare股份公司总裁兼首席执行官)
翻译|项安波(石药集团) 董方(东方伊诺医疗科技)
中文版翻译负责人|医药研发达人主编 高野哲臣
• ICH-GCP于1997年在日本以GCP省令颁布翌年实施,J-GCP虽然反映ICH-GCP但仍有差异,特别是研究机构负责人被赋予了过多职责,以及各研究机构各自制定知情同意书(ICF, Informed Consent Form)模版等,构成了日本特有的现象。
• J-GCP体制下,与ICH-GCP相比,由于大量文件需以研究机构负责人为收发对象,造成申办者、伦理委员会(IRB, Institutional Review Board)、主要研究者与研究机构负责人之间不必要文件往来。这只是其中一个例子,笔者呼吁对日本当前不合理、非高效、不够迅速、过于注重质量、成本高昂、且落后于时代发展的临床试验制度进行根本性变革。
• R&D Head Club 制定日本 ICH 通用模板,日本制药工业协会(JPMA)修订后,厚生劳动省推荐并呼吁各方积极采用。笔者向包括中国在内的海外制药企业建议:今后,在日本开展临床试验时积极采用JPMA制定的ICF通用模板。
本系列A《日本的临床试验和药品市场》第四篇,由高野向大家介绍“ICH-GCP与J-GCP的区别”,以及由此差异所催生的“日本的知情同意书(ICF, Informed Consent Form)通用模板”。
需要注意的是,本文多处亦包含笔者的主观观点,敬请读者留意。
01 ICH-GCP与J-GCP的区别
如第1期、系列A第一篇的《日本临床试验的历史(上)》中所述,ICH E6(GCP)于1996年6月10日达到E6(R1)的第4阶段,并于1997年3月在日本作为GCP省令(新GCP)颁布,1998年4月全面实施。
这部J-GCP(GCP省令,新GCP)虽然是为了反映ICH-GCP而制定的,但正如图1所示,从1997年颁布起直至今天,ICH-GCP与J-GCP之间依然存在差异。由这些差异所引发的问题——例如,各家研究机构各自设立伦理委员会(IRB, Institutional Review Board)并自行制定知情同意书(ICF)模板——可以说是日本特有的现象,但笔者认为,这已成为当今日本出现药品错失问题和药品上市延迟问题再度加剧的原因之一。
在ICH-GCP制定之前,日本自1990年10月起就实施了局长通知(旧GCP),因此GCP省令(新GCP)在基本反映ICH-GCP的同时,仍以延续局长通知(旧GCP)的形式于1997~1998年公布及实施。
如前所述,ICH-GCP与J-GCP之间存在差异。例如,在J-GCP框架下,不允许申办者(Sponsor)与主要研究者直接签署合同,而仅允许申办者与研究机构签署合同。
此外,原则上要求各实施临床试验的研究机构必须各自设立IRB,且向IRB提出审查申请的主体不是主要研究者,而是该研究机构的负责人(如院长)。
J-GCP还进一步规定,研究机构的负责人须承担选任从事临床试验行政工作的事务人员的职责,即设立“临床试验办公室”。(图1)
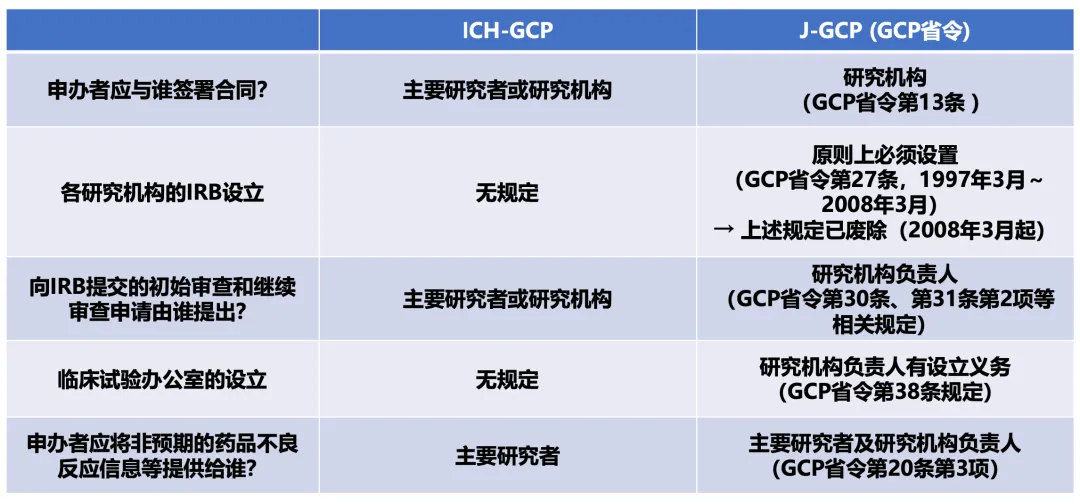
图1. ICH-GCP与J-GCP(GCP省令)的主要区别(节选)
(高野哲臣,t2T Healthcare Inc.,2025年4月20日制作)
根据PMDA的公开信息(https://www.pmda.go.jp/review-services/trials/0008.html),截至2025年3月31日,日本共有1,299家临床试验的IRB。虽然各实施临床试验的研究机构原则上必须设立IRB的规定,已在GCP省令颁布11年后的2008年3月颁布GCP省令修订被废除(见图1),但从其后日本IRB的数量并未出现大幅减少来看,可以推测,那些在2008年3月之前就已设有院内IRB的研究机构,并未积极转向“废除院内IRB → 利用院外IRB”的模式。关于日本的临床试验IRB的实际情况,将在系列A的下一篇第五篇中进行详细阐述。
此外,J-GCP(GCP省令)中从临床试验申办到合同签署的流程(图2)显示,日本的J-GCP体制下,由于研究机构负责人承担着极为重要的职责,因此有很多必须以研究机构负责人为收件人和发件人的文件。然而,在日本,围绕慢性疾病(如生活习惯病)开展的临床试验,往往是在个人诊所(Clinic)中进行,而诊所的“主要研究者 = 研究机构负责人”。因此,从图2中的⑤“临床试验的指示与决定通知书”等文件开始,从临床试验启动前一直到试验结束/中止后,在主要研究者与研究机构负责人之间会存在许多不必要的文件往来。
再者,由于J-GCP中赋予研究机构负责人的职责(图1),也导致在临床试验期间与安全性信息相关的IRB审查次数和文件处理量明显高于美国。
(https://www.jpma.or.jp/information/evaluation/symposium/Rinsho_202502_Soukai.html)
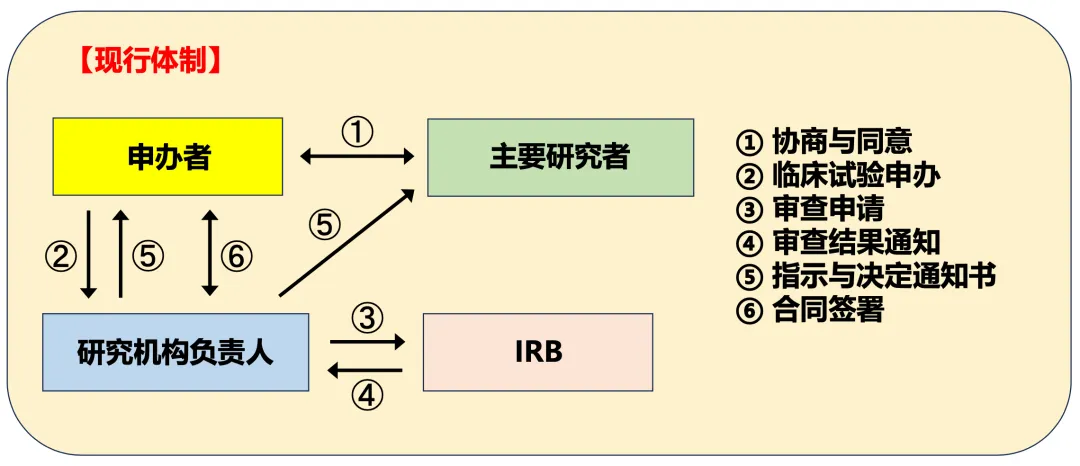
图2. 日本J-GCP中从临床试验申办到合同签署的流程【现行体制】
(引自“厚生劳动省的第12届关于临床试验运作方式研究会,资料2,2007年2月28日,东京”,部分改编)
此外,如图3所示,日本制药工业协会(JPMA)早在2007年时就已提出建议:“应将临床试验中研究机构负责人的多数职责转移至主要研究者,如ICH-GCP中设定的那样,相应的权利与义务应向主要研究者集中。”然而,该建议至今尚未被落实。
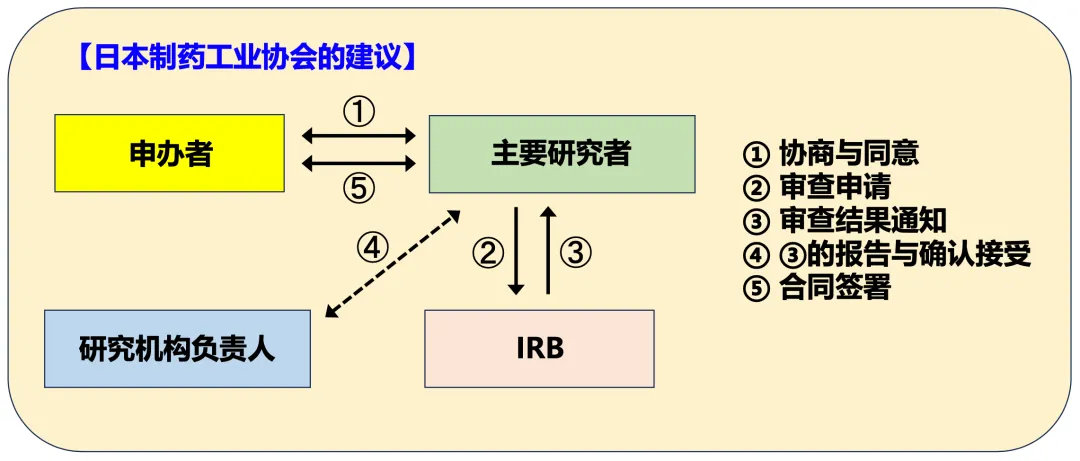
图3. J-GCP中从临床试验申办到合同签署的流程【日本制药工业协会(JPMA)的建议】
(引自“厚生劳动省的第12届关于临床试验运作方式研究会,资料2,2007年2月28日,东京”,部分改编)
目前,ICH-GCP(E6)正在进行全面修订,其最新版本E6(R3)的原则(Principles)及附件1(Annex 1)已于2025年1月6日达到第4阶段,而E6(R3)的附件2(Annex 2)也已于2024年11月至2025年3月进行公开征求意见。
随着E6(R3)的推行,全球范围内的临床试验将迅速向包括去中心化临床试验(DCT, Decentralized Clinical Trials)、实效性随机对照试验(Pragmatic Clinical Trials)以及真实世界数据(Real-World Data)等在内的新型临床试验模式推进。
为了响应E6(R3),日本厚生劳动省(MHLW)在2025~2026年间肯定会对J-GCP(GCP省令)进行全面修订。如果不借此机会对日本当前不合理、非高效、不够迅速、过于注重质量、成本高昂、且落后于时代发展的临床试验制度进行根本性变革,那么笔者认为日本所面临的药品错失与药品上市延迟问题今后也无法得到实质性改善。
02 各研究机构自有的知情同意书(ICF)模板
接下来,将围绕由ICH-GCP与J-GCP差异所引发的问题之一——各研究机构自有的ICF模板进行说明。
在日本开展临床试验的研究机构中,许多医院拥有各自的模板用于知情同意书(ICF),并要求在院内的所有临床试验中使用该模板来制作ICF,笔者考虑原因如下:
无论是哪个科室的哪项临床试验,都必须由研究机构负责人(院长)向院内IRB提交审查申请,且研究机构是临床试验合同的签署者。因此,无论是从院长、院长管理下的临床试验办公室还是院内IRB的立场出发,始终使用研究机构自有的ICF模板,更有利于确认GCP合规性、补充设施要求等方面的管理。
始终使用研究机构的自有模板可确保内容格式、逻辑顺序和术语表达的一致性,因而临床试验办公室在准备IRB审查资料时能更高效地进行事前确认,院内IRB委员也更易于审查,主要研究者(PI)/次级研究人员(Sub-I)与临床研究协调员(CRC)也能更顺利地开展临床试验参与者的知情同意说明工作。
主要研究者作为研究机构的一员,如不使用已经获得研究机构负责人(院长)、临床试验办公室和院内IRB共识的模板,而试图以其他模版制作的ICF来进行临床试验,也是不现实的。
尽管申办者通常会提供ICF草案,但其模板因申办者而异。如前所述,日本多数实施临床试验的研究机构会要求在提交IRB前,将申办者的模板转换为研究机构的自有模板。这往往意味着CRA(临床监查员)和CRC需要花费数小时乃至十几小时对每个申办者的ICF进行替换和内容修改,同时还需在申办者内部进行该版本ICF的公司层级审批,这一流程可能需耗时几天至两周。这样的流程在每一项临床试验中、每一家研究机构中都会被重复执行,从而在全国范围内累积产生庞大的资源消耗与人力成本。
(https://www.jpma.or.jp/information/evaluation/results/allotment/q83i5d0000000plj-att/CL_202406_material.pdf)
值得一提的是,尽管GCP规定的必需要素都被涵盖,但由于各研究机构的ICF模板存在差异,即使是同一临床试验,不同的研究机构向临床试验参与者提供的信息(解释内容)也会有所不同。笔者认为,从临床试验参与者的角度来看,这种状况绝非理想。
03 日本ICH通用模板的制定、发布与使用推广
解决各研究机构自有ICF模板所带来的问题,使研究机构与申办者实现双赢的,是新开发的日本ICH通用模板。
该模板由一个日本制药企业研发部门负责人组成的自发性组织——R&D Head Club(https://rdhead-club.com/)于2021年左右开始着手制定,名称为Common ICF Template,于2022年10月4日发布了Ver.1版本,并于2023年3月14日发布了Ver.1.1版本。此后,由JPMA对该模版进行了进一步的修订,使其具有更高的通用性,直至今天。
JPMA制定的ICF通用模板,在整体结构及对临床试验的一般性说明内容方面设定为不可更改,同时对于临床试验特有内容也规定了一定的填写规范,从而确保其作为全国统一模板的意义不被削弱,实现对临床试验参与者信息的统一,同时达到减轻研究机构及申办者在ICF的制作、审查和同意说明工作方面的负担。
值得注意的是,由JPMA制定的这份ICF通用模板,在第3期图4中“日本药品监管部门应对药品错失和药品上市延迟的措施示例”一栏所提及的 “7. Common ICF Template”,已被MHLW在2024年7月4日以医政局研究开发政策课长及医药局药品审查管理课长发布的通知(医政研发0704第1号、医药药审发0704第2号)《关于在临床试验中使用知情同意书通用格式的利用推荐通知》(https://www.mhlw.go.jp/content/10800000/001272350.pdf)明确指出,其内容“符合GCP省令等相关规定”,并广泛呼吁研究机构、制药企业、CRO、SMO等积极采用。
JPMA对该2024年7月4日MHLW通知的英文翻译版链接如下:
https://www.jpma.or.jp/information/evaluation/results/allotment/q83i5d0000000plj-att/CL_202411_Notification_en.pdf
有关ICF通用模板的JPMA官方资料链接整理如下:
JPMA药品评审委员会 临床评价分会的说明(2025年4月)
https://www.jpma.or.jp/information/evaluation/results/allotment/CL_202406_material.html
知情同意书(ICF)通用模板(第1.2版、2025年3月28日)
https://www.jpma.or.jp/information/evaluation/results/allotment/q83i5d0000000plj-att/CL_202503_ICF_ver.1.2.docx
知情同意书(ICF)通用模板(第1.2版、2025年3月28日)[参考英文翻译版]
https://www.jpma.or.jp/information/evaluation/results/allotment/q83i5d0000000plj-att/CL_202503_ICF_ver.1.2_en.docx
知情同意书(ICF)通用模板 说明资料
https://www.jpma.or.jp/information/evaluation/results/allotment/q83i5d0000000plj-att/CL_202406_material.pdf
知情同意书(ICF)通用模板 宣传手册
https://www.jpma.or.jp/information/evaluation/results/allotment/q83i5d0000000plj-att/CL_202408_material.pdf
知情同意书(ICF)通用模板 宣传手册[参考英文翻译版]
https://www.jpma.or.jp/information/evaluation/results/allotment/q83i5d0000000plj-att/CL_202411_Material_en.pdf
截至2025年4月15日,已有包括以下所列企业在内的30家制药企业导入了ICF通用模板。与此同时,越来越多的研究机构也在推进其使用。
旭化成制药、Aska Pharmaceutical、安斯泰来制药、艾伯维、EA制药、大冢制药、MSD、Kissei Pharmaceutical、杏林制药、葛兰素史克、盐野义制药、生化学工业(Seikagaku Corporation)、Zeria Pharmaceutical、大正制药、大鹏药品工业、武田药品工业、田边三菱制药、帝国制药、日本勃林格殷格翰、拜耳药品、辉瑞、富士胶片富山化学、丸石制药(Maruishi Pharmaceutical)、丸保(Maruho)、杨森制药
最后,笔者希望包括中国在内的外国制药企业,今后在日本开展临床试验时,能够积极采用JPMA制定的ICF通用模板。
综上,围绕“ICH-GCP与J-GCP的差异”,以及“日本的ICF通用模板”进行了介绍。为了解决日本的药品错失与药品上市延迟问题,笔者认为修订J-GCP中不合理和非高效的部分,以及推广ICF通用模板是非常重要的。
下一篇(系列A第五篇)将介绍:关于【日本药品监管部门应对药品错失和药品上市延迟的措施示例】中的【日本的临床试验IRB,例如中心伦理(*)的推进】等内容。(请参见第3期文章中的图4第5项)
(*)中心伦理:每个临床试验只有一个IRB
 产业资讯
瞪羚社 2026-06-18
367
产业资讯
瞪羚社 2026-06-18
367
 产业资讯
深蓝观 2026-06-18
383
产业资讯
深蓝观 2026-06-18
383
 产业资讯
研发客 2026-06-18
427
产业资讯
研发客 2026-06-18
427
 热门资讯
热门资讯 微信公众号
微信公众号