

 产业资讯
产业资讯
 药创新
药创新  2025-07-26
2025-07-26
 1960
1960
这场风波将整个基因治疗领域推入对安全与监管的深刻拷问。
美国明星生物技术公司正深陷一场前所未有的监管风暴。2025年7月中旬,3例患者死亡阴影笼罩下,FDA要求Sarepta暂停其临床试验,同时要求其明星产品Elevidys自愿停止发货,Sarepta竟强硬拒绝。然而,仅72小时后,公司戏剧性宣布:全面暂停Elevidys发货。这场风波将整个基因治疗领域推入对安全与监管的深刻拷问。
Sarepta再掀波澜
2025年7月18日,美国FDA发布公告,鉴于3例可能与公司产品有关的死亡事件,对Sarepta Therapeutics针对肢带型肌营养不良症(LGMD)的在研基因疗法临床试验实施临床暂停。同时,撤销Sarepta的AAVrh74平台技术认定。
FDA领导层与Sarepta会面,要求其当日自愿停止Elevidys的所有发货,然而,该公司拒绝执行这一要求,理由是自己有对数据的全面科学解释。但是,2025年7月21日,Sarepta却宣布将在7月22日营业结束时暂停Elevidys的所有发货。
这一系列消息在基因治疗行业和患者群体中引发轩然大波。Sarepta公司一直致力于罕见病基因疗法的研发,而Elevidys更是其明星产品,此前已在市场上引起广泛关注。如今,这些事件的发生,让人们对基因疗法的安全性和监管问题产生了深深的忧虑。
Elevidys作为全球首款杜氏肌营养不良(DMD)基因治疗药物,自诞生起便备受瞩目。2023年6月,它获得FDA加速批准,定价高达320万美元(约合人民币2300万元)一个疗程,成为全世界第二贵的药。2024年6月,其适用人群年龄扩大到4岁及以上。
然而,这款药从获批开始就争议不断。它没有硬性疗效证据,通过FDA加速审评程序上市。在FDA的专家咨询委员会会议上,该药的专家投票为8票赞成、6票反对。
审批依据的临床试验未直接设置疗效终点,而是以“患者骨骼肌中观察到了截短型抗肌萎缩蛋白的表达”这一替代终点作为审批参考。反对的专家们认为,这无法证明其与患者临床症状的改善有关。进一步分析显示,Elevidys对4-5岁儿童的行走能力有一定提升,但对6-7岁儿童并无显著效果,这也是FDA当时设置严格年龄限制的原因。
从市场表现看,Elevidys是Sarepta的核心营收来源。公司披露,2025年第二季度Elevidys净产品收入达2.82亿美元,占公司总净产品收入5.13亿美元的55%以上,足见其对公司的重要性。
作为首款获批的杜氏肌营养不良基因疗法,它在市场上曾占据先发优势。获批初期,因针对罕见病且治疗需求迫切,市场需求旺盛,尤其在扩大适应证至不能行走的患者后,进一步拓宽了市场覆盖。但随着安全事件频发,其市场前景蒙上阴影,股价暴跌,投资者对其市场潜力的信心受挫。
最近的2例Elevidys相关患者死亡事件均为急性肝衰竭导致。接连的死亡事件,让Elevidys的安全性受到严重质疑。尽管急性肝损伤是Elevidys及其他腺相关病毒(AAV)介导的基因疗法已知的潜在副作用之一,且已在处方标签中予以明确说明,但如此严重的不良事件发生,还是让患者、家属以及医疗界感到不安。
面对FDA的要求,Sarepta陷入了两难境地。7月16日,公司宣布已依据FDA要求为Elevidys补充针对急性肝损伤和急性肝衰竭的黑框警告。同时,计划裁员约36%,并暂停多个药物的研究,以节约成本应对危机。
基因疗法的全球现状
自2012年欧洲批准Glybera以来,基因治疗行业开始高速发展。据Fortune Business Insights分析,2019年全球基因治疗市场规模为36.1亿美元,预计到2027年将达到356.7亿美元。批准的基因治疗产品数量也不断增加,涉及肿瘤、遗传性疾病等多个领域。
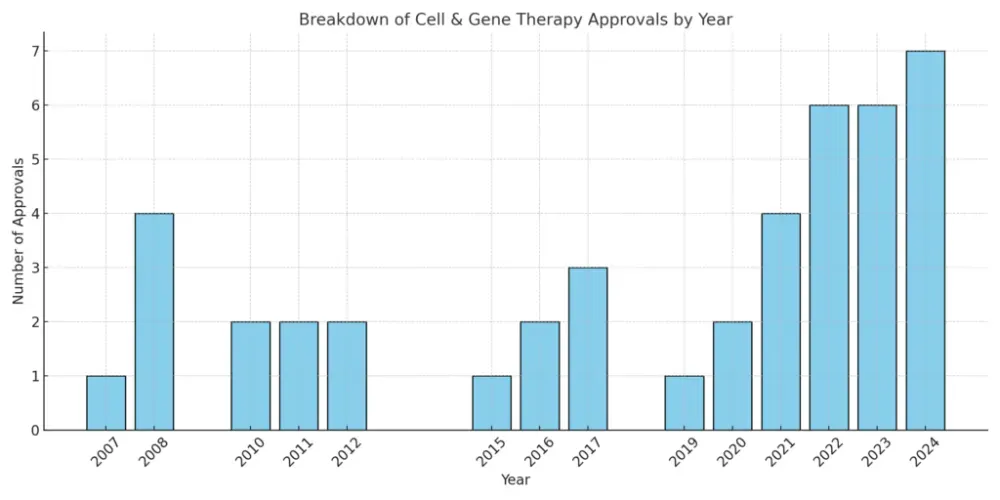
各年FDA细胞和基因疗法批准数量/图源:bioinformant.com
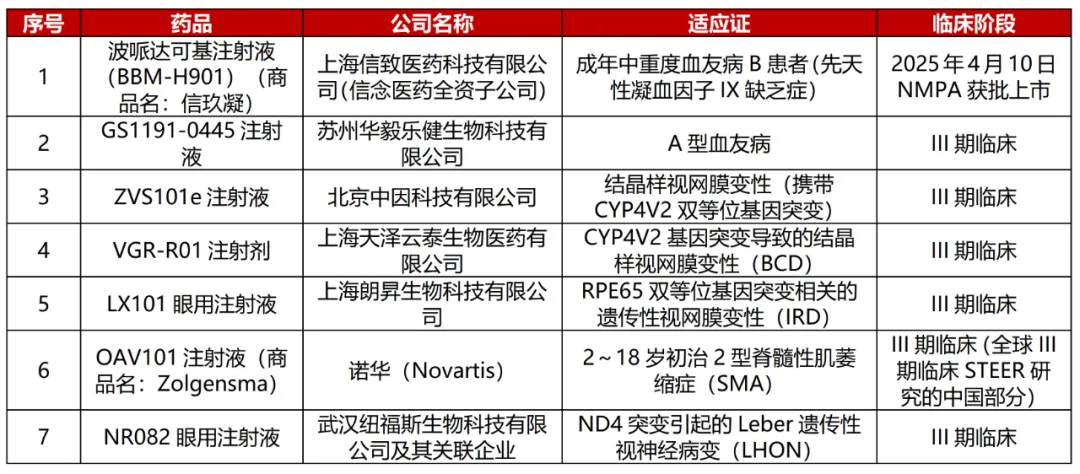
国内AAV基因疗法盘点(1款上市、6款进入III期临床、43款IND获批)/数据来源:《国内AAV基因疗法盘点》报告
基因疗法虽然在研发上取得了一定的进展,但商业化之路却异常艰难。高昂的研发成本导致基因疗法定价极高,从100万美元到350万美元一针不等,如此“天价”让全世界的卫生系统都难以负担。
辉瑞的B型血友病基因疗法Beqvez,2024年4月获FDA批准,定价350万美元,上市后因支付谈判、患者筛选流程等原因,尚无有商业保险覆盖的患者完成治疗。罗氏的遗传性视网膜疾病基因疗法Luxturna,市场表现远低于预期,2024年销售额约合2000万美元,同比下降了59%。
即使像诺华的Zolgensma表现不错(2024净销售额12.14亿美元),历史上也出现过安全事件;Sarepta的Elevidys刚带领公司迈过盈利线,就因安全问题陷入困境。
药企虽一直在探索支付创新,如分期付款等模式,但美国医保方(Medicaid)并不支持,商业保险也因“一次性治疗无法纳入年费模型”而拒付。新兴市场医保覆盖有限,导致基因疗法的可及性非常低。
近年来,跨国大药企(MNC)在基因疗法领域的态度发生了巨大转变。2025年2月,辉瑞宣布终止B型血友病基因疗法Beqvez的研发与商业化,并清空所有基因治疗管线,彻底退出该领域。10年间,辉瑞在基因治疗领域投入大量资金,却未获得满意的回报。
今年3月,罗氏宣布对旗下基因治疗公司Spark Therapeutics启动“根本性重组”,计提了高达24亿美元的商誉减值损失。Spark曾凭借全球首款遗传性视网膜疾病基因疗法Luxturna成为行业标杆,但其市场表现不佳,多个管线项目被砍。
挑战与希望并存的未来
Sarepta的事件为整个基因治疗行业敲响了警钟,安全性和疗效的平衡、商业化的困境以及监管的挑战,都是基因疗法发展道路上必须克服的难题。Elevidys出现的急性肝损伤等副作用,凸显了AAV载体技术的局限性,这也促使行业加快对新型载体的研发,例如探索更安全的病毒载体改造或非病毒载体技术,以降低潜在风险。
从监管层面看,FDA对Sarepta平台技术认定的撤销,以及对临床试验的严格管控,有利于推动监管体系进一步完善。未来或许会建立更精细化的风险评估机制,在加速罕见病药物审批的同时,加强对长期安全性的监测。
支付方面,“天价”基因疗法的可及性问题亟待解决。Sarepta等企业探索的支付创新虽遇阻,但也为行业提供了经验。未来可能需要政府、医保、企业多方协作,建立可持续的支付体系,例如基于疗效的分期付款、医保专项基金等,让更多患者用得起这一突破性疗法。
从全球范围看,基因治疗临床试验的数量持续增长,美国和中国在这一领域积极探索,主导了大量试验,为疗法的优化和改进积累了宝贵数据。并且,随着科技的飞速发展,新型载体和基因编辑技术不断涌现,有望攻克现存难题。
尽管当前基因疗法面临诸多挑战,但对于DMD等罕见病患者而言,它仍是重要的希望。随着技术的不断进步、监管的日益完善以及支付模式的创新,基因疗法有望逐步克服困难,真正实现从实验室到临床的跨越,为更多患者带来福音。
 产业资讯
瞪羚社 2026-06-18
443
产业资讯
瞪羚社 2026-06-18
443
 产业资讯
深蓝观 2026-06-18
462
产业资讯
深蓝观 2026-06-18
462
 产业资讯
研发客 2026-06-18
505
产业资讯
研发客 2026-06-18
505
 热门资讯
热门资讯 微信公众号
微信公众号