特别评述 | 人工智能助力药物研发:可解释性深度神经网络分子表征模型
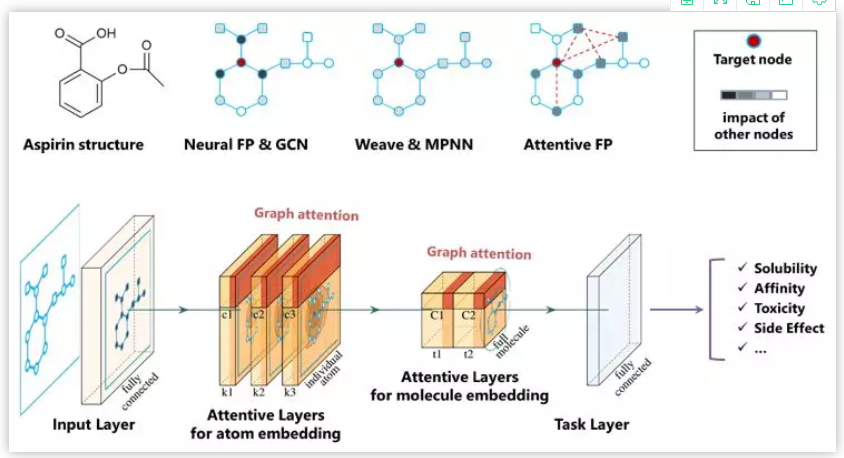
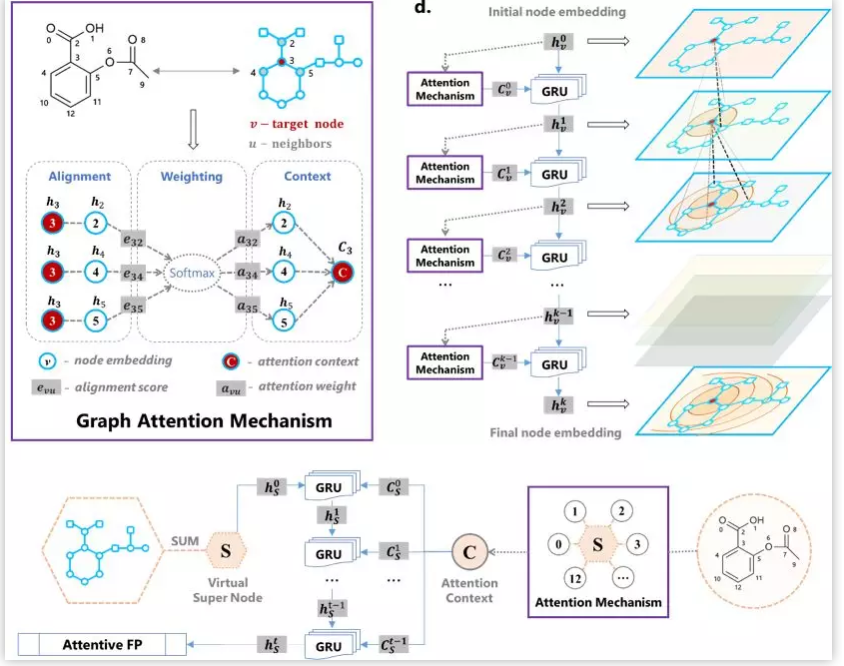
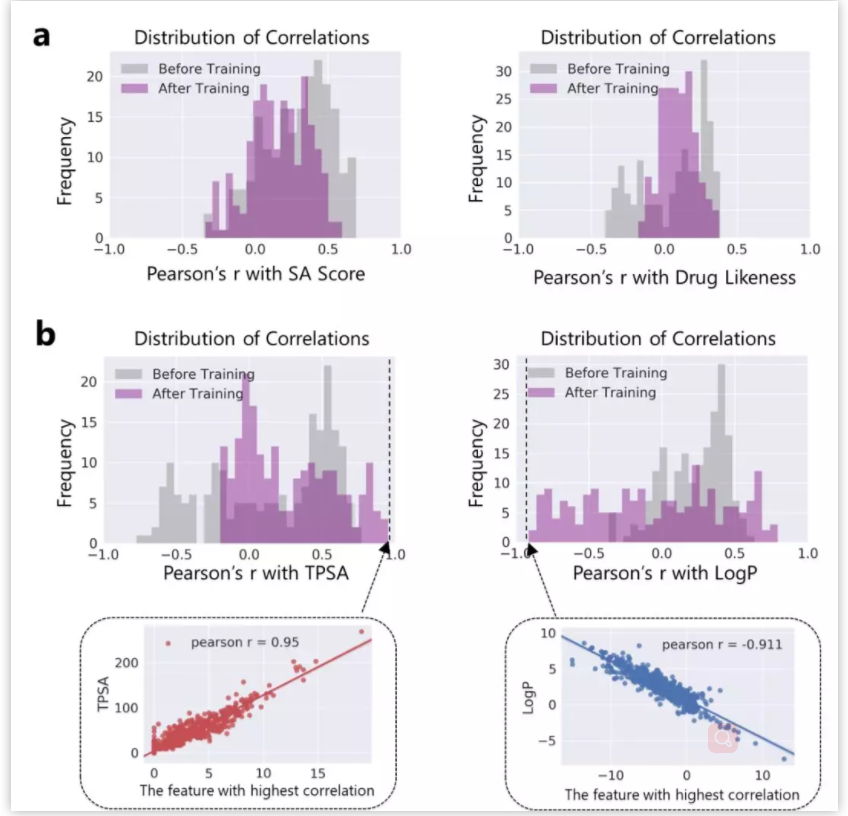
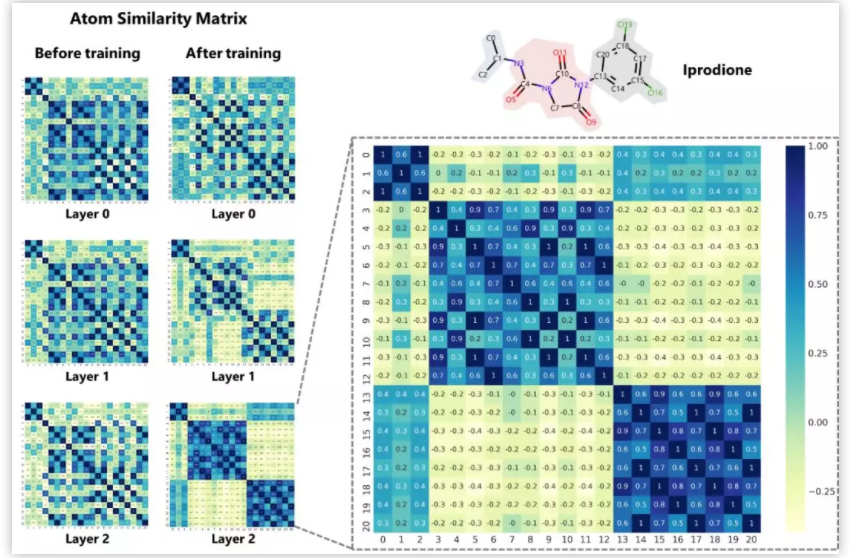
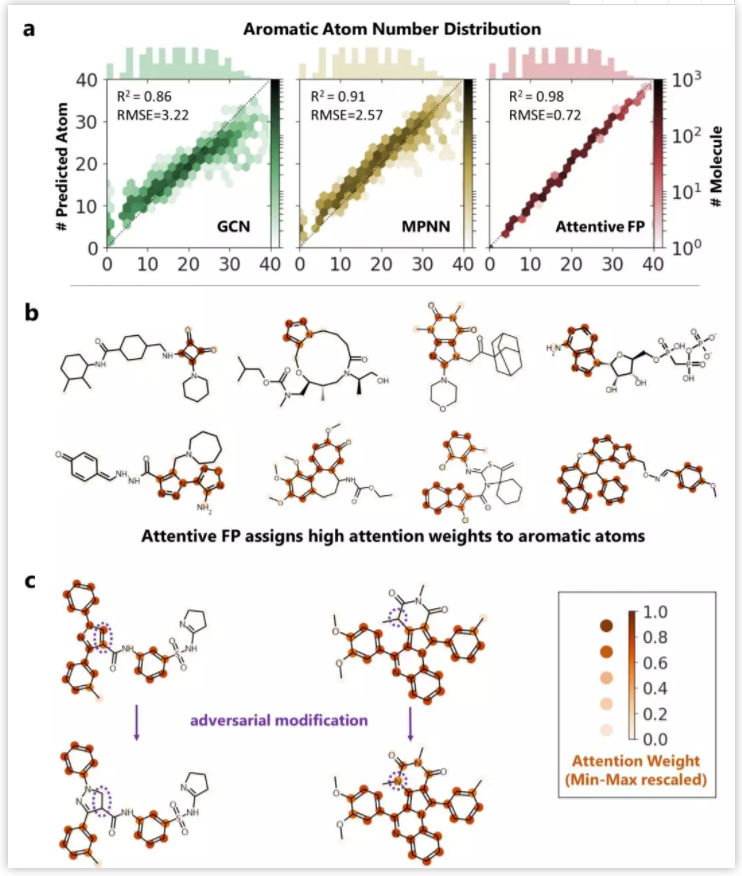
生命科学迅猛发展,刷新大家对生命认知的同时,也给疾病治疗带来了更多的可能性。理论上,几乎所有生物学功能都可以被药物靶向。小分子因其相对低廉的成本,成为各大制药公司和研究机构开展疾病治疗研究的首选工具,药物发现也因此越来越“平权化”。然而,找到具有合适药理学、毒理学和药代动力学等特性的小分子,依然是一个很大的挑战。面对不断增长的药物研发数据,现有的人工智能 (AI) 方法虽然可以据此构建出强大的预测模型,然而深度神经网络所学到的东西通常难以被化学家和生物学家理解,而且这种认知差距正在不断增大,也使科技人员难以相信AI模型的预测结果,这也限制了AI技术在药物研发实践中的应用 【1】 。 近日,中科院上海药物所蒋华良 院士和郑明月 研究员 (第一作者为博士研究生熊招平 ) 在Journal of Medicinal Chemistry Pushingthe Boundaries of Molecular Representation for Drug Discovery with the GraphAttention Mechanism 。 该论文介绍了一种基于注意力机制的图神经网络模型 (Attentive FP) 。该模型可以用于分子表征,在多个药物发现相关的数据集上的预测表现达到当前最优,并且该模型所学到的内容具有可解释性。 这种可解释性在机器的认知和人的认知的差异间架起了一座桥梁,由此可能更好地利用机器的认知增强药物学家的认知,产生更大的实际应用价值。 Attentive FP的特征可视化表明,它可以自动从特定任务中学习到分子结构内非局部的特性,因此可以帮助药物学家或化学家超越经验和直觉,直接从各种性质数据中获取对该分子结构更深层的理解。 结构决定性质,性质体现于结构。如何从一个分子的化学结构中提取出它的各种性质是科学家梦寐以求的目标。到目前为止,人们先后发明了5000种以上的不同描述符 (特征) 去表征一个化学分子的结构 【2】 。传统的机器学习模型就围绕这些预定义的描述符,通过特征工程选取不同的组合,对小分子的各种性质进行建模预测。特征工程选取是一个繁琐且耗时的过程,而且这种较强的预设先验很可能使模型产生偏差,导致预测效果达不到最优。以Neural FP为代表的分子图神经网络模型能以较少的特征描述符作为输入,得到明显更优的预测结果,是人工智能在分子表征领域的重要尝试 【3】 。然而,在机器学习中准确性和可解释性很难兼顾。如果不能使神经网络的“黑盒子”透明化,人们很难判断一个模型只是拟合或记住了训练数据,还是真正具备了泛化的能力。面对药物研发后期巨大的成本投入,药物学家不可能完全相信某个黑盒算法给出的“武断”预测 【4】 。因此,人工智能药物设计研究的重点之一就是需要探索深度学习算法的可解释性,针对性地开发了更符合化学背景,更易于解读的人工智能模型。 图1. Attentive FP总体框架以及与同类的图神经网络模型比较。 作者比较了他们自己的AttentiveFP模型与其他几种图神经网络模型 (图1) 。如果将分子看作为一张图 (graph) ,给定一个节点 (红色标记的目标原子) ,在Neural FP 【3】 和GCN 【5】 模型中,其他节点对目标节点的影响会随距离严重衰减,这不符合化学直觉,即化学结构中距离较远的原子间有时也会产生较强的影响,比如分子内氢键的形成;Weave 【6】 和MPNN (特指Deepchem中实现的MPNN) 模型则默认所有其他节点对目标节点有相同的影响,这可以更好捕捉分子结构中的一些非局部特征,但显然忽视了化学分子固有的结构。作者提出的Attentive FP能在保持分子固有结构的情况下,有效捕捉图的非局部特征和远距离节点相互作用 (图2) 。这得益于Attentive FP先在原子水平加入注意力机制,学习到分子的局部特征,后在整个分子水平加入注意力机制,学习到分子的全局特征。值得一提的是,相比其他同类图神经网络模型,Attentive FP用了最少的初始特征作为模型输入,依然在多个测试数据集中达到了当前最优的预测表现。 图2. Attentive FP 注意力图神经网络架构。 图3 AttentiveFP学习水溶性时自动学习到的特征与化学家定义的描述符比较。 作者将Attentive FP学习水溶性时自动学习到的特征与化学家定义的描述符进行了对比 (图3) 。可以看到,训练前后,自动学习到的特征几乎能复现出跟预测任务相关的经验描述符。这些经验描述符是根据化学家的专业知识所定义出来的,比如TPSA (拓扑分子极性表面积) 和LogP (油水分配系数) 。因为这些经验描述符与水溶性高度相关,对预测任务有较强的指导,传统的机器学习模型通常会直接选择这些描述符作为输入特征的一部分来预测水溶性。但作者发展的Attentive FP可以不以这些化学先验作为输入,而以更原始简单的输入 (如原子和键的类型等) ,直接在在隐含层中自动学习到的这些人们长期积累的化学知识,表现为训练后的模型隐含层特征与这些经验描述符的相关性变高,而与预测任务不相关的描述符如SA Score (合成难易性) 和Drug Likeness (类药性) 与学习到的隐层特征相关性比较并没有显著变化。 图4. Attentive FP自动学习化学环境。 作者发展的AttentiveFP模型还能自动学习到原子所处的化学环境,比如,以药物溶解度作为监督任务进行训练,将模型学习到的原子向量作相似性评估,负相关的原子对标为黄色,正相关的标为蓝色 (图4) 。结果表明通过学习,整个分子显示出特定的结构模式,这种模式在隐藏层的高层更加明显。对于图4所示的Iprodione结构,原子被自动聚集的三个部分正好对应分子结构中的三个片段,其中,分子结构中灰色背景的化学基团极性比较小,不利于水溶性,中间红色背景的基团极性较大,利于水溶性。这一结果提示模型可能自动学习了到各个原子所处的不同化学环境。 图5. Attentive FP自动学习到分子中的芳香性子结构。 化学分子中的芳香性是一种典型的非局部特征,这一问题也给基于卷积架构的图神经网络模型带来了挑战。通过利用注意力机制,作者发展的Attentive FP模型很好的解决了这一问题。 当原始输入中去除编码芳香性的相关特征(避免信息泄露) ,输出仅以分子中芳香原子的个数作为学习目标进行监督训练时,Attentive FP能根据注意力机制的权重,准确标出芳香原子的位置。同时,训练完成的模型面对对抗性的样本 (微小的结构改变,但对芳香性影响巨大的分子) 也能进行准确鉴别,展现了强大的泛化能力。 人工智能在人脸识别、语音识别、翻译和自动驾驶等方面的应用不需要关注智能算法学习到了什么,为什么会做出这样的判断,只要达到足够的精度即可。但对于像药物发现这种科学问题,其中有更多的不确定性,在通用人工智能把整个新药发现流程包办以前,药物学家会更相信自己的经验直觉,但同时又希望从越来越多的药物研发数据中汲取新的见解。数据的积累和深度学习算法的应用可以建立更准确的预测模型,而这些预测如果是不能被解释,或者说被药物学家理解,那么将很难取得药物学家的信任,进而被真正应用而成为药物发现必不可少的环节。 该团队开发的基于注意力机制的 可解释 图神经网络分子指纹Attentive FP是对人工智能的可解释性在药物发现中的有益探索,它将机器认知与人的认知连接起来,以期更好地利用机器的认知增强药物学家的认知,这类前沿且与药物研究需求紧密结合的探索,相信能产生更大的实际应用价值。 整体来说,文章干货满满,更多内容可自行查阅原文,有兴趣的读者也可以直接利用公开的代码做自己的探索, https://github.com/OpenDrugAI/AttentiveFP 。 值得一提的是,蒋华良/郑明月 课题组前不久也在Journal of Medicinal Chemistry 杂志上发表人工智能助力药物研发的论文,根据现有激酶活性大数据,应用深度神经网络算法,建立了药物调控激酶谱的预测分析方法 (详见此前BioArt的报道: 特别评述 | 人工智能助力药物研发:深度学习预测药物调控激酶谱 ) 。 吴朝晖 (浙江大学校长,中国科学院院士,人工智能研究专家) 评论家布雷特·金 (Brett King) 在《智能浪潮: 增强时代来临》 一书中指出,今天所探索的人工智能等科技,将彻底重新定义人类的下一个时代,这一时代可称之为智能增强时代。智能增强时代不可避免地要协调好机器智能和人类智能的关系,在决策中融合机器智能,实现人机协同,增强人类智能。 最近十年,得益于算力的增长和数据的累积,我们注意到深度学习在物流、监控、个人助手、高频交易等领域取得了突破性的成功,推动了这一波的人工智能热潮。然而,我们同样发现目前以深度神经网络为代表的连接主义人工智能 (AI) 还有很多局限:它太依赖于数据,欠缺泛化推理能力,也是人类不能理解的“黑箱”。我们不知道神经网络得到预测的依据,更不确定人工智能模型究竟是学习到了可泛化的知识,还是仅仅记住了样本,拟合了数据。对于诸如图像识别、机器翻译等任务,有些情况下即使产生错误并不会产生严重后果,只要模型达到好的预测效果,我们可以不必关心模型是如何做出预测的。有很多人工智能任务,会因为人类对智能系统理解不足,而存在的巨大风险。深度神经网络如果一直保持“黑箱”状态,模型会很容易受对抗性样本的攻击,用户也很难决定什么时候可以信任模型的预测。例如,医生不知道AI模型对病理图片作出预测的依据,就不能放心采用AI给出的诊断结论;药物学家不知道AI系统为什么优选开发某些分子而不是另外一些分子的原因,面对后期巨大的经费投入和失败风险,就很难相信AI给出的研发决策。 因此,当前人工智能想要真正显示智慧特征,创造普惠价值,需要解决的一个重要问题是深度神经网络的可解释性。只有人工智能决策过程变得更加透明,这种智能才能更加通用。可解释性人工智能 (Explainable AI) 作为一个非常前沿的研究方向,聚焦于用系统性和可解释的方式呈现人工智能所学习到的复杂逻辑,让人工智能的预测依据更好地被人类理解。这是实现人机协作,增强人类智能的基础。可解释性人工智能受到了各国政府、工业界和学术界的广泛关注。美国国防部先进研究项目局DARPA资助了可解释性人工智能项目XAI (ExplainableAI) ;中国国务院在2017年印发的《新一代人工智能发展规划》中提出要“实现具备高可解释性、强泛化能力的人工智能”,得到了产业界和学术机构广泛认可和积极响应。 近日,上海科技大学和中科院上海药物研究所蒋华良院士和郑明月研究员团队在人工智能的可解释性在药物发现领域中的应用进行探索,开发了一种新的分子结构表征方法Attentive FP 【8】 。该方法是基于注意力机制的图神经网络模型。其中,图神经网络模型对拓扑图中的点和边的关系进行建模,有利于关系推理。注意力机制是近年来人工智能自然语言模型的核心进展之一,在提高语言模型性能的同时也提升了模型的可解释性 【9】 。在Attentive FP中,该团队使用图神经网络处理含有原子和键的分子图结构,并通过创新性地引入原子水平和分子水平的注意力机制,使分子图模型兼具推理能力和可解释性。利用Attentive FP进行分子表征和药物性质预测建模,可以获得泛化性能更好的模型;通过可视化模型自动学习到的特征,可以发现Attentive FP能从化合物的性质数据中直接学习和提取符合化学经验知识的模式和关联性。这些特性可以帮助药物学家更加高效地处理日益扩增的研发数据,从海量大数据中直接获取新的见解,丰富药物学家的知识库和经验储备。 未来,随着脑科学、认知科学、类脑计算的迅猛发展,人工智能在感知、记忆、推理等方面的功能“短板”终将得到补齐,人工智能“黑箱”模型也将变得更加透明,届时人工智能才能更显智慧特征,更具普惠价值。可以预见,药物研发领域也会融合机器智能与人类智能,实现人机协同,由机器智能帮助药物学家快速处理海量数据,增强药物研发的合理决策。基于注意力机制的图神经网络分子指纹Attentive FP是在药物发现中对可解释性人工智能的积极尝试,也展示了可解释性人工智能在人机协同以及在助力药物研发方面的巨大潜力。
作者简介
熊招平 ,上海科技大学与中科院上海药物研究所联合培养博士生,导师为蒋华良院士和郑明月研究员。他研究兴趣集中于图神经网络(Graph Neural Network)在分子表征、分子生成和结构优化中的应用,重点探索可解释性人工智能在新药研发中的潜力。他思维活跃,研究兴趣广泛,对多学科交叉融合尤其感兴趣。2015年第二届中美青年创客大赛中,他所在的无人船团队获得上海赛区第一名,他在其中负责视觉算法开发和布署。2016年第一届由中科院微小卫星中心举办的微小卫星设计大赛中,他主持的微小卫星生化实验模块项目获得第二名。2018 DREAM Challenge的“多靶点药物预测挑战赛”(Multi-Targeting Dream Challenge 2018) 中,他在甲状腺髓样瘤(medullarythyroid carcinoma)和tau蛋白神经退行性模型两项任务都获得第一名(人工智能助力上海研究生熊招平摘取多靶点药物分子设计国际挑战大赛冠军 )。
郑明月 ,中国科学院上海药物研究所研究员、博士生导师、国家新药研究重点实验室成员、中国化学会计算机化学专业委员会委员。研究方向是基于人工智能和大数据的精准药物设计技术开发。在药物作用机制和靶点发现、新靶点活性化合物的发现和成药性优化等方面取得了一系列成果,发展了具有特色和创新性的机器学习算法和模型,得到了国内外同行的关注。近年来,共发表SCI论文70余篇,参与5部专著的编写;在Trends Pharmacol Sci、Autophagy、J Med Chem、J Chem Theory Comput和Bioinformatics
蒋华良 ,中国科学院院士,中国科学院上海药物研究研究员,上海科技大学免疫化学研究所教授。1987年毕业于南京大学化学系,获得有机化学学士学位;1992年于华东师范大学化学系获得物理化学硕士学位;1995年于中国科学院上海药物研究所获得药物化学博士学位。蒋华良长期致力药物科学基础研究和新药发现,他通过生物学、化学、数理科学和计算信息科学等多学科的交叉,开展原创药物研究新策略与新方法、先导化合物发现和优化、药物靶标调控机制等研究。他发展了一系列靶标发现和药物设计新方法,被国际同行和制药公司广泛应用。他发展了能预测化合物药效的理论计算方法,部分解决了药物设计领域的重大难题。他针对多种重要靶标发现了数十个新结构类型的先导化合物,其中5个候选药物已进入临床试验研究,。迄今他在国际学刊上发表论文460余篇,其中通讯或共同通讯论文作者200余篇、综述13篇;合编专著24本,译著2本,论著被他引2万余次。申请专利160项,获授权70余项(其中国际专利16项),实现成果转让6项。 他获国家自然科学二等奖、国家科技进步二等奖、何梁何利科技进步奖、国家青年科学家奖、国家青年科技奖、上海市牡丹自然科学奖、上海市科技进步一等奖、上海市科技精英等多种奖项。目前担任J. Med. Chem.副主编和其他5种国际学刊的编委。曾任国家863计划专家组成员、国家重大基础研究计划“蛋白质科学重大基础研究计划”专家组成员、国家自然科学基金委“基于化学小分子探针的信号转导过程研究”重大研究计划专家组成员,现任国家自然科学基金委“生物大学分子动态修饰与化学干预”重大研究计划专家组组长。 https://doi.org/10.1021/acs.jmedchem.9b00959
1. Shake-up in AI drug discovery, NatBiotechnol 2. Dragon 7.0. https://chm.kode-solutions.net/products_dragon.php 3. Duvenaud, D.; Maclaurin, D.;et al.. Convolutional Networks on Graphs for Learning Molecular Fingerprints.ArXiv E-Prints 4. Schneider, G. Mind and Machinein Drug Design. Nat. Mach. Intell. 5. Zhou, Z.; Li, X. GraphConvolution: A High-Order and Adaptive Approach. ArXiv E-Prints 6. Kearnes, S.; McCloskey, K.;Berndl, M.; Pande, V.; Riley, P. Molecular Graph Convolutions: Moving BeyondFingerprints. J Comput-Aided Mol Des 7. Wu, Z.; Ramsundar, B.; et al..MoleculeNet: A Benchmark for Molecular Machine Learning. Chem Sci 8. Xiong, Z, Wang, D,; et al..Pushing the boundaries of molecular representation for drug discovery withgraph attention mechanism. J Med Chem. 9. Ashish, V.; Shazeer N,; et al.."Attention is all you need." In Advances in neural informationprocessing systems,
phirda@phrda.com
010-58156160
回到顶部


 研发追踪
研发追踪
 BioArt
BioArt  2019-09-08
2019-09-08
 14086
14086


 热门资讯
热门资讯 微信公众号
微信公众号