

 产业资讯
产业资讯
 研发客
研发客  2022-02-28
2022-02-28
 6168
6168
在姚晨教授看来,美国FDA这次对数据造假的提法无论对中国药监还是临床研究行业,都是一次警醒。FDA通常会考量研究项目之前是否接受过FDA对临床试验数据的核查,国内临床研究领域以后需加大注意。国家药监局或者在将来可以更加系统化地定期披露临床试验现场核查质量和数据,制定年报。

近日,美国FDA肿瘤药物咨询委员会(ODAC)经讨论投票,最终以14:1的投票结果支持FDA的提议:由信达生物/礼来的PD-1抗体信迪利单抗用于联合培美曲塞和铂类化疗用于非鳞状非小细胞肺癌一线治疗的生物制品申请许可(BLA),需要补充更多的临床试验数据,以证明该药在美国患者人群和美国医疗实践中的适用性。
这件事引起了制药界的广泛关注。除了关于临床试验设计中,单一中国数据是否符合美国医疗实践、与FDA的沟通交流以及临床终点设定等核心问题的讨论,FDA 在ODAC上还引用了《英国医学杂志》(BMJ)在2016年发表文章的表述:“调查发现80%的中国临床试验数据有欺诈性”。

这一公开指责令姚晨教授等国内众多学者非常痛心。日前,研发客专访了北京大学第一医院医学统计室主任、北京大学临床研究所副所长姚晨教授,请他谈一谈对这次美国FDA重提中国数据造假的看法。在姚晨教授过去6年以来的观察中,中国药监部门和临床研究学者为保障数据的真实性和完整性开展了哪些工作?我国的临床试验数据质量是否有实质性提高?
姚晨教授是我国为数不多的临床研究方法学专家,他精通临床医学、生物统计学、流行病学和循证医学等等学科。2018年,姚晨与北京大学临床研究所阎小妍、董冲亚等针对2016年BMJ发表的新闻报道,撰写了观点文章Protecting the accuracy clinical data in China(《保障中国临床试验数据的准确性》),并在线发表在BMJ网站上。
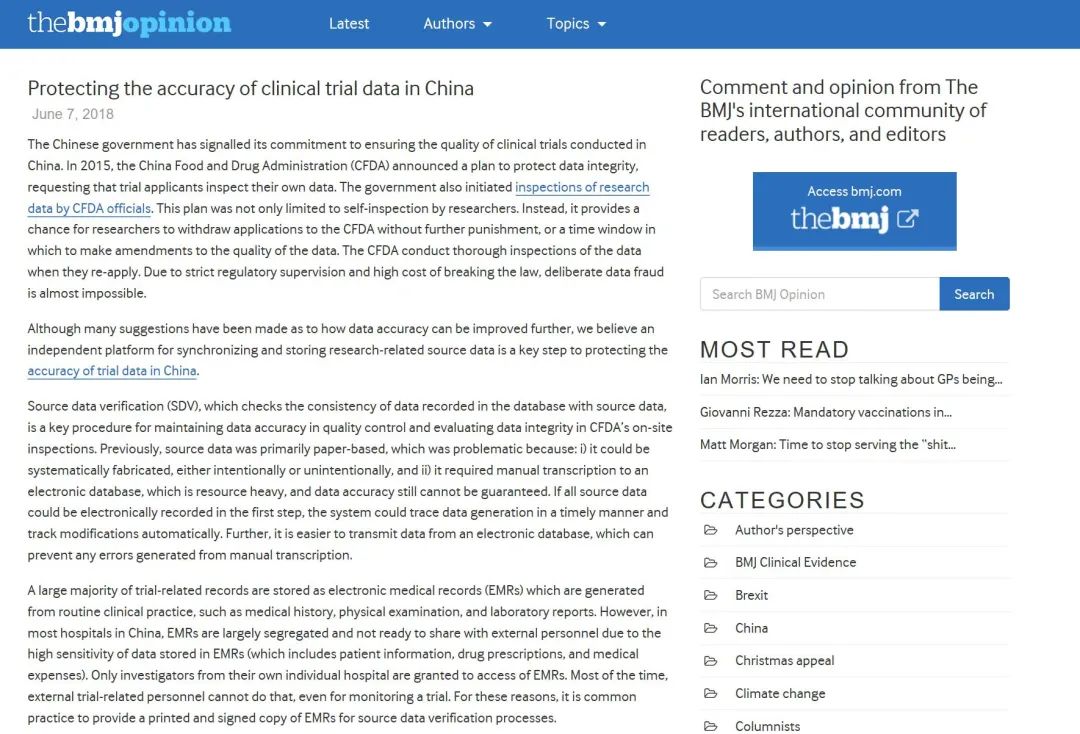
2018年6月,姚晨团队在BMJ在线发表观点文章。
在此短文中,他首先提到了原国家食品药品监督管理局(CFDA)在2015年7月22日,发布的《关于开展药物临床试验数据自查核查工作的公告》j(2015年117号)(下称“722公告”), “722公告”给予申办方自查核查的机会,可在不受进一步严厉处罚的情况下撤回药品注册申请,或提供改善数据质量的时间窗。截止到2015年底,在1622个药品注册申请受理号中,有超过80%的申请被要求撤回进行自查。因此,BMJ新闻稿中以此“80%的中国临床试验数据有欺诈性”作为吸引眼球的标题是不妥的。实际上“722公告”也是我国临床研究监管的分水岭标志。原CFDA审核查验中心(CFDI)启动了对研究数据的专项检查,并在此次专项检查之后,将基于风险对药品上市注册研究进行常规检查成为常态,“溯源”、核查原始数据成为研究者、申办者、监管机构对于研究数据质量眼中的重中之重。
“四个最严”
在姚晨教授看来,中国临床试验数据质量自2016年以来,已发生了可喜的变化。“722公告”发布后,中国药监部门以最严谨的标准、最严格的监管、最严厉的处罚、最严肃的问责(业内俗称的“四个最严”)为监管工作的根本遵循和行动指南,出台系列法律法规,强化对注册申请的现场检查,尽最大所能保证在我国开展临床试验的质量。
2017年6月19日,原CFDA正式加入国际人用药品注册技术协调会(ICH),成为其全球第8个监管机构成员。这意味着我国药品监管部门、制药行业和研发机构,将逐步转化和执行国际制药研发生产的最高技术标准和指南。
2020年7月1日起施行的新版《药物临床试验质量管理规范》(Good Clinical Practice, GCP)要求研究者应当确保所有临床试验数据是从临床试验的源文件和试验记录中获得的,是准确、完整、可读和及时的。源数据应当具有可归因性、易读性、同时性、原始性、准确性、完整性、一致性和持久性。
2021年9月,为进一步优化我国临床研究规范管理举措、促进临床研究创新积累经验,国家卫健委还正式发布了《医疗卫生机构开展研究者发起的临床研究管理办法(试行)》。办法指出:“医疗卫生机构应当建立临床研究源数据的管理体系,实现集中统一存储,保障临床研究数据在收集、记录、修改、处理和保存过程中的真实性、准确性、完整性、规范性、保密性,确保数据可查询、可溯源。”
“一系列组合拳下,中国的临床试验数据从标准、监管以及处罚问责三个方面大幅提高了要求。与此同时,药厂、CRO和临床研究机构投入大量的人力和技术,紧绷了质量这根弦。由于国家对临床试验造假零容忍,加之严格的监管和高昂的违法成本,蓄意的数据欺诈已几乎不可能了。”姚晨说。
突破源数据壁垒
早在2018年BMJ在线发表的观点文章里,姚晨教授团队就指出,“源数据”是临床试验质量问题的症结所在,并首次提出了保障中国临床研究源数据质量的具体解决方案。
由于医院信息系统中的电子健康记录(Electronic Medical Records, EMRs)并非为临床研究设计和服务的,EMR中涵盖大量的敏感信息,医院很难单独授权医院外部人员直接访问这部分电子化源数据,用来进行临床试验的源数据核查,也难以提供数据传输接口将EMR数据直接传输至第三方的临床试验数据采集系统中。
因此,目前的常规操作还是将电子化的EMR打印签字作为纸质源数据溯源使用。同时这部分电子数据打印出后还需要进行人工转录至临床试验数据采集系统。这种落后的纸质源数据采集流程操作繁琐,且数据准确性、可溯源性难以得到保证,也容易产生漏洞。
姚晨教授指出,进一步提高我国临床研究数据质量的核心环节在于,推进临床研究源数据的电子化。建议在医院信息中心建立独立的电子化临床研究源数据平台,一方面可以将临床研究涉及的电子病历(EMR)数据同步存储至该平台,一方面可以整合随访等临床试验特定的随访数据收集,这样临床试验涉及的源数据都以电子化源数据形式整合至该平台。从而在根源上回避了纸质源数据在收集、整理、核对、溯源检查上的缺陷。
2019年5月起,国家药品监督管理局发布一系列真实世界证据支持药品、医疗器械研发和审评相关指南。同时,海南博鳌乐城作为临床真实世界数据试点工作开展。其中,所有指南提及对于利用真实世界证据产生的真实世界证据源数据提出了明确的质量和溯源要求。
基于该设想,姚晨教授团队结合多年临床研究的实践,满足上述政策法规对于临床研究数据采集的要求,以真实世界数据采集和治理作为突破口,将当年的设想初步落地——与博鳌乐城临床研究中心和杭州莱迈医疗信息科技有限公司合作开发了创新型电子源数据记录(ESR,eSource Record)工具。并在《中国食品药品监管》上发表的《真实世界数据采集、治理与管理的一体化解决工具研究》文章中揭示了ESR工具研发的探索过程。

姚晨教授表示,令人欣慰的是,2022年2月8日,国家卫健委就十三届全国人大四次会议第10294号建议答复称“国家卫健委高度重视国家健康医疗大数据服务,正在研究建立全国统一的电子健康档案、电子病历、药品器械、公共卫生、医疗服务、医保等信息标准体系,并逐步实现互联互通、信息共享和业务协同。”这意味着,我国从前限制临床研究源数据电子化采集的壁垒即将被打破。
是诟病,也是警醒
近年来,越来越多我国自主研发或与跨国制药公司合作开发的新药走出国门。同时,中国作为重要的临床研究中心,参与了大量的国际多中心临床试验,开展的临床研究数据质量有目共睹。
“尽管信达/礼来的研究由于人群代表性等原因未能支持用于直接境外上市申请,但如果仅基于一篇2016年理解片面的新闻报道,就否定我国近年来临床研究数据质量,这是错误的。”姚晨教授说。
在他看来,美国FDA这次的提法无论对中国药监还是临床研究行业,都是一次警醒,FDA通常会考量研究项目之前是否接受过FDA对临床试验数据的核查,因此,国内临床研究从业者以后需加大注意。同时,国家药监局或可更加系统化地定期披露临床试验现场核查质量和数据,制定年报,使整个行业能从具体的质量缺陷案例中吸取教训,更好提高中国临床试验质量。
未来,提高中国临床试验机构、研究者和研究的水平和声望,让世界了解中国的研究者,信任中国的临床试验,是今后一段时间中国临床试验研究者职责和使命。“相信在监管部门和临床研究学界、业界的努力下,中国临床试验研究数据质量将继续稳步提升。”姚晨教授说。
参考文献:
FDA Raises Concerns About China Developed Drugs
1.https://www.wsj.com/articles/fda-raises-concerns-about-china-developed-drugs-11644408180
2. Woodhead, Michael. 80% of China's clinical trial data are fraudulent, investigation finds[J]. Bmj, 2016:i5396.
3.Protecting the accuracy of clinical trial data in China
https://blogs.bmj.com/bmj/2018/06/07/protecting-the-accuracy-of-clinical-trial-data-in-china/
4. 国家食品药品监督管理总局关于开展药物临床试验数据自查核查工作的公告(2015年第117号)
https://www.nmpa.gov.cn/xxgk/ggtg/qtggtg/20150722173601172.html
5. 国家食品药品监督管理总局成为国际人用药品注册技术协调会成员
https://www.nmpa.gov.cn/zwgk/xwfb/20170622153001224.html
6.关于公开征求《真实世界证据支持药物研发的基本考虑》意见的通知
https://www.cde.org.cn/main/news/viewInfoCommon/7e6fb9fc3f066a966a02130f24dbff1c
7.关于公开征求《用于产生真实世界证据的真实世界数据指导原则(征求意见稿)》意见的通知
https://www.cde.org.cn/main/news/viewInfoCommon/794a436a118459d867f8f2ac95edfd85
8. 国家药监局关于发布真实世界数据用于医疗器械临床评价技术指导原则(试行)的通告(2020年第77号)
https://www.nmpa.gov.cn/xxgk/ggtg/qtggtg/20201126090030150.html
9. 关于医疗卫生机构开展研究者发起的临床研究管理办法(征求意见稿)公开征求意见的公告
http://www.nhc.gov.cn/qjjys/s7945/202012/630fa2bf316d48a4856f8727450c429b.shtml
10. 对十三届全国人大四次会议第10294号建议的答复
http://www.nhc.gov.cn/wjw/jiany/202202/043135aa8fd8405c9c1b6a008a5bf5cd.shtml
11. 姚晨,谢红炬,郝新宝等.真实世界数据采集、治理与管理的一体化解决工具研究[J].中国食品药品监管,2021,(11):62-70.
 产业资讯
药智网 2026-06-10
443
产业资讯
药智网 2026-06-10
443
 产业资讯
恒瑞医药 2026-06-10
397
产业资讯
恒瑞医药 2026-06-10
397
 产业资讯
氨基观察 2026-06-10
386
产业资讯
氨基观察 2026-06-10
386
 热门资讯
热门资讯 微信公众号
微信公众号