

 产业资讯
产业资讯
 医药时间
医药时间  2024-06-06
2024-06-06
 709
709
据不完全统计,2024年1-5月,FDA拒批13款药物,涉及自身免疫性疾病、神经系统疾病、肿瘤、抗体药物、免疫疗法等多个方面。
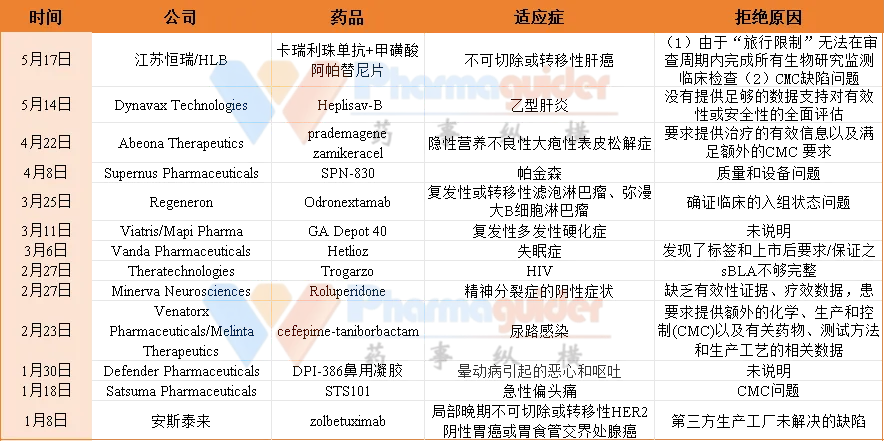
安斯泰来:zolbetuximab
2024年1月9日,安斯泰来宣布其收到FDA就CLDN18.2单抗zolbetuximab上市申请发出的完整回复函(CRL),暂时拒绝批准上市,作为Claudin18.2阳性、HER2阴性的局部晚期不可切除或转移性胃或胃食管交界处(G/GEJ)腺癌的一线治疗方案。

近日,安斯泰来宣布,美国食品药品监督管理局(FDA)确认该公司zolbetuximab的生物制品上市许可申请(BLA)的重新递交。
此项递交的BLA是基于GLOW 和SPOTLIGHT 两项III期临床试验的结果。SPOTLIGHT试验评估了zolbetuximab联合mFOLFOX6(一种包括奥沙利铂、亚叶酸和改良的5-氟尿嘧啶的联合化疗方案)相比于安慰剂联合mFOLFOX6的疗效。GLOW试验评估了zolbetuximab联合CAPOX(一种包括卡培他滨和奥沙利铂的联合化疗方案)相比于安慰剂联合CAPOX的疗效。
在GLOW 及 SPOTLIGHT两项临床试验中,根据经验证的免疫组织化学分析判定,参加试验的肿瘤患者中,约38%的患者符合CLDN18.2表达的阳性标准(定义为≥75%的肿瘤细胞中显示中度至强的CLDN18膜染色)。
如获批上市,zolbetuximab将成为美国首款针对CLDN18.2患者群体的靶向治疗选择。
Satsuma Pharmaceuticals:STS101
2024年1月18日,FDA 拒绝批准日本SNBL旗下Satsuma Pharmaceuticals用于治疗急性偏头痛的双氢麦角胺鼻粉产品STS101上市。STS101 是鼻腔给药偏头痛药物甲磺酸双氢麦角胺 (DHE) 的重新配制版本,利用 Satsuma 的专有设备进行递送,整体只有口红大小,方便携带和使用。
完整回应函 (CRL) 中指出了STS101的化学、制造和控制 (CMC) 方面的问题。CRL 没有指出 STS101 有任何安全问题,也没有要求进行额外的研究。Satsuma 将与 FDA 讨论拒绝事宜,以重新提交上市申请。

Defender Pharmaceuticals:DPI-386鼻用凝胶
1月30日,Defender Pharmaceuticals宣布FDA签发了一份完整答复函(CRL),拒批了该公司用于预防晕动病引起的恶心和呕吐的鼻内东莨菪碱凝胶(DPI-386鼻用凝胶)NDA。该公司没有说明监管机构拒绝该药物的原因。

Minerva Neurosciences:Roluperidone
2月27日,Minerva Neurosciences宣布收到FDA就Roluperidone用于治疗精神分裂症阴性症状的新药申请(NDA)发出的完整回复函(CRL),表示拒批其精神分裂症新药。

在CRL中,FDA 指出了以下四点拒批理由:
虽然MIN-101C03研究在主要疗效终点上显示出统计学意义,但其本身不足以确立实质性的有效性证据;
提交的NDA缺乏有关同时服用抗精神病药物的数据;
NDA材料缺乏证明Roluperidone对改善精神分裂症阴性症状具有临床意义的必要数据;
在已提交的安全性数据中,至少服用12个月拟议剂量(64mg)Roluperidone的患者数量不足。
为了解决这些缺陷,FDA规定Minerva必须至少再提交一项积极、充分和对照良好的研究,以支持Roluperidone治疗阴性症状的安全性和有效性。Minerva还必须提供额外的数据来证明Roluperidone与其它抗精神病药物联合用药的安全性和有效性,以进一步支持Roluperidone的有效性并证明拟议剂量的长期安全性。
Theratechnologies:Trogarzo
2月27日,Theratechnologies(纳斯达克股票代码:THTX)宣布,美国食品和药物管理局(FDA)已就该公司关于一种长效、CD4导向、附着后HIV-1抑制剂Trogarzo®(ibalizumab-uiyk)维持剂量的肌肉注射(IM)给药方法的补充生物制品许可申请(sBLA)发出了拒绝备案函(RTF)。

经初步审查,FDA认定该sBLA不够完整,无法进行实质性审查。RTF指出,sBLA没有包含建立Trogarzo® IM给药途径与静脉输注给药途径之间药代动力学桥梁所需的数据。
Venatorx/Melinta:cefepime-taniborbactam
2月23日,Venatorx Pharmaceuticals和Melinta Therapeutics宣布,FDA发布了关于头孢吡肟-他尼波巴坦(cefepime-taniborbactam)新药申请(NDA)的完整回应函(CRL)。CRL没有发现NDA中的临床安全性或有效性问题,FDA也没有要求任何新的临床试验来支持头孢吡肟-他尼波巴坦的批准。FDA要求提供额外的化学、生产和控制(CMC)以及有关药物、测试方法和生产工艺的相关数据。

Regeneron:Odronextamab
3月25日,再生元Regeneron宣布,FDA拒绝其CD3/CD20双抗Odronextamab治疗复发性或转移性滤泡淋巴瘤(FL)和弥漫大B细胞淋巴瘤的上市申请,再生元表示唯一原因是确证临床的入组状态问题,FDA在完整的回复函(CRL)中没有发现任何与有效性、安全性、试验设计、标签或制造有关的问题。

Viatris/Mapi Pharma:GA Depot
3月11日,晖致(Viatris)和Mapi Pharma共同宣布收到FDA就GA Depot(醋酸格拉替雷,40mg)用于治疗复发型多发性硬化症的新药申请(NDA)发出的完整回复函(CRL)。
关于FDA拒绝的具体细节很少,这两家公司仅表示,他们目前正在审查完整回复函,以更好地确定GA Depot 40的“适当下一步”。

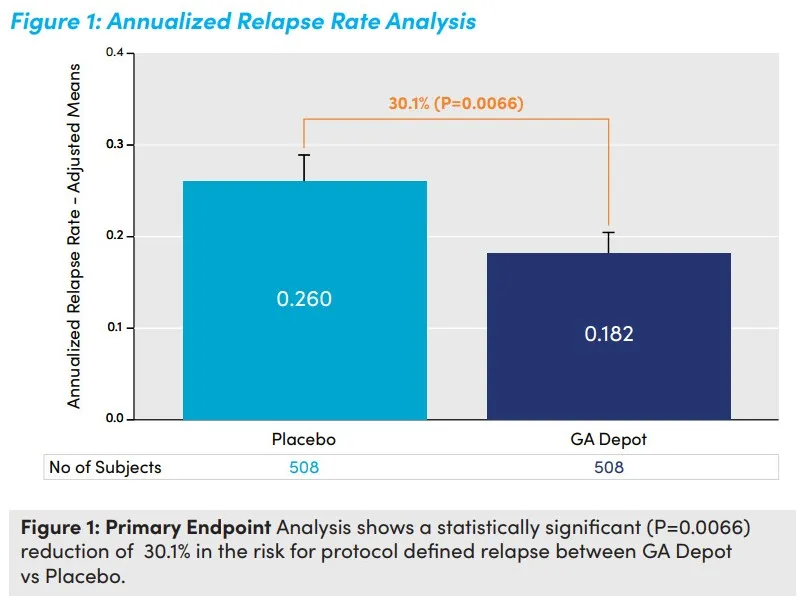
GA Depot的NDA主要是基于一项III期研究的积极结果。该研究共纳入1016例患者,评估了GA Depot对比安慰剂治疗RMS患者的有效性、安全性和耐受性。结果显示,GA Depot组患者的年复发率相比安慰剂组降低了30.1%(P=0.0066)。
Vanda Pharmaceuticals:Hetlioz
3月6日,Vanda Pharmaceuticals宣布,已经收到FDA就Hetlioz(他司美琼)治疗失眠补充新药申请(sNDA)发出的完整回应函。
FDA表示由于发现了标签和上市后要求/保证之外的缺陷,该公司针对Hetlioz的补充新药申请(sNDA)无法以目前的形式获得批准。
Vanda Pharmaceuticals表示将进一步评估FDA的回复,对其进行审查,并考虑未来的行动方向。

Supernus:SPN-830
4月8日,FDA第三次拒绝了Supernus Pharmaceuticals的帕金森治疗药物SPN-830的新药上市申请,该监管机构不认为SPN-830以目前的形式做好上市的准备了。
CRL中表示有两个方面需要FDA进行更多审查。
第一个问题是Supernus需要提供更多信息,包括有关产品质量的更多信息。Supernus指出最近向监管机构提交了更多的产品质量数据,但尚未得到审查。
FDA 提出的另一个问题与输液设备的主文件有关。Supernus 表示,主文件是专有的,并计划与设备制造商讨论监管机构要求的信息和重新提交 NDA 所需的步骤。与此同时,Supernus公司表示,没有发现任何临床安全性或有效性问题需要获得批准。
Abeona:prademagene zamikeracel
4月22日,Abeona Therapeutics宣布:其细胞疗法 prademagene zamikeracel (pz-cel,EB-101) 收到了 FDA 的完整回复信 (CRL),FDA拒绝了该疗法上市,并要求其提供治疗的有效信息,以及满足额外的CMC 要求才能批准申请。
Abeona需要满足FDA对生产以及释放测试的要求,以确保药品的纯度和安全性。
对此,Abeona向FDA提交了计划,承诺在BLA批准之前提供CMC数据,并在2024年中期获得批准后提供完整的验证报告。随后在非正式会议上,Abeona与FDA进行讨论。
根据FDA发出的完整回复函(CRL),FDA表示,Abeona提交数据的拟议时间不允许FDA在2024年5月25日PDUFA日期之前完成对数据的审查,现在将有所推迟。
CRL没有发现任何与BLA中的临床疗效或安全性数据相关的缺陷,FDA也没要求任何新的临床试验或临床数据来支持pz-cel的批准。
实际上,pz-cel的被拒早有苗头。2023年8月,Abeona在与FDA的BLA前会议上,FDA还要求Abeona提交的BLA材料中包含额外的背景和数据,以及CMC数据和临床主题的补充数据。
Dynavax:Heplisav-B
5月14日,Dynavax Technologies 宣布 FDA 拒绝了其重组蛋白乙型肝炎疫苗用于在血液透析中患者的补充生物制品许可证申请。
这家加州生物制药公司表示,它收到了监管机构关于其sBLA的完整回复函(CRL),其中包括来自119名接受血液透析的成人的四剂量Heplisav-B方案的I期HBV-24研究的临床免疫原性和安全性数据。根据CRL,“申请没有提供足够的数据来支持对有效性或安全性的全面评估。”

FDA 认为该申请没有提供足够的数据来支持有效性或安全性的全面评估。CRL表示,HBV-24的数据不足,因为第三方临床试验现场运营商销毁了大约一半参加试验的受试者的数据源文件。该公司还透露,FDA发现单臂研究中的受试者总数“不足以评估四剂方案的安全性”。
江苏恒瑞/HLB:“双艾”疗法
5月17日,恒瑞医药发布公告称,其收到美国食品药品监督管理局(FDA)关于PD-1抑制剂卡瑞利珠单抗联合阿帕替尼用于不可切除或转移性肝细胞癌患者的一线治疗的上市申请的完整回复信,FDA需要全面评估企业对生产场地检查缺陷的答复。同时由于部分国家的旅行限制,FDA表示在审查周期内无法全部完成该项目必需的生物学研究监测计划临床检查。
对此,恒瑞表示将尽快重新提交申请。

这两款药物的中文商品名称都有艾字,所以恒瑞医药称其为“双艾”组合疗法。因为肝癌患者群体大,且申请上市的是一线治疗方案,所以“双艾”组合疗法在美国的上市是恒瑞医药的工作重点。
2023年1月,“双艾”组合疗法的上述适应症在中国获批。2023年10月,恒瑞医药把“双艾”组合疗法许可给韩国上市公司HLB的美国子公司Elevar Therapeutics,后者获得“双艾”组合疗法用于治疗肝癌适应症除大中华区和韩国以外的全球范围内开发和商业化权利。
参考资料:各公司官网、生物药大时代、药事纵横
 产业资讯
新药说 2025-06-16
98
产业资讯
新药说 2025-06-16
98
 产业资讯
Medaverse 2025-06-16
96
产业资讯
Medaverse 2025-06-16
96
 产业资讯
猎药人俱乐部 2025-06-16
101
产业资讯
猎药人俱乐部 2025-06-16
101
 微信公众号
微信公众号 热门资讯
热门资讯 热点标签
热点标签