

 产业资讯
产业资讯
 病有所依
病有所依  2025-01-18
2025-01-18
 1643
1643
近年来,随着快速审批政策的实施和医保目录常规化调整,抗肿瘤药物的研发与应用迎来重要机遇与挑战。文章总结了2024年下半年新获批抗肿瘤药物及其关键性临床试验的特点,并基于国外卫生技术评估(Health Technology Assessment,HTA)机构的推荐意见,提出了关于创新抗肿瘤药物医保准入的建议,为各相关方提供参考。
一、新获批抗肿瘤药物基本情况
在2024年下半年,共统计到23个肿瘤领域的新获批适应症(对应23个抗肿瘤药物),其中43.5%(10/23)的药物为国产药物。从申报情况来看,其中82.6%(19/23)的药物为首次申请上市,17.4%(4/23)的药物为增加新适应症;从适应症来看,69.6%(16/23)的药物新批准适应症为实体瘤,30.4%(7/23)的药物为血液瘤,其中排名前四位的癌种分别为肺癌(5个)、淋巴瘤(4个)、白血病(2个)、前列腺癌(2个)和胃癌(2个)。获批新适应症癌种分布见图1。
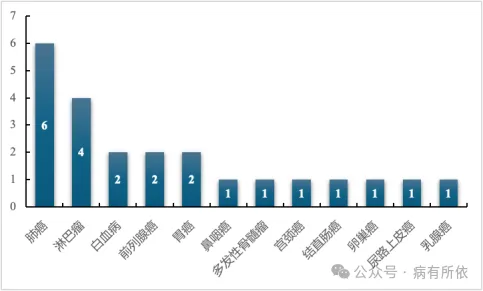
图1 抗肿瘤药物获批新适应症癌种分布
从作用机制来看(见图2),超过半数(52.2%,12/23)为靶向药物,其次为抗体药物偶联物(ADC)(包括替朗妥昔单抗、芦康沙妥珠单抗、索米妥昔单抗和维恩妥尤单抗4个药物),免疫检查点抑制剂(包括莫妥珠单抗、纳武利尤单抗、艾帕洛利托沃瑞利单抗和塔戈利单抗4个药物),CAR-T细胞疗法(包括瑞基奥仑赛和西达基奥仑赛2个药物)和1个化疗药物(紫杉醇口服溶液)。
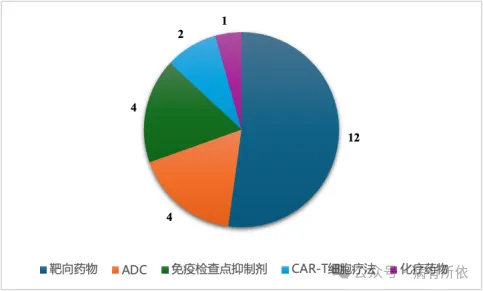
图2 抗肿瘤药物作用机制分布
从靶点来看,多个新靶点迎来创新药。维恩妥尤单抗是国内首个被批准上市的Nectin-4 ADC药物,也是迄今为止全球范围内唯一一款上市的Nectin-4 ADC;氟泽雷塞由劲方医药自主研发,是国内第一种获得批准的KRAS G12C抑制剂,打破了过去四十年KRAS突变NSCLC患者面临的“有靶无药”的困境,填补了临床空白;芦康沙妥珠单抗由科伦博泰研发,是国内首个获批、全球第二个获批上市的TROP2 ADC药物。
在本次统计的样本中,除了多样的作用机制和创新靶点外,还涉及到医保准入的热点问题。例如,紫杉醇口服溶液作为全球首款口服紫杉醇药物,其市场定位和医保准入策略备受关注。目前,医保目录中已收录4种紫杉醇制剂:纳入常规目录的紫杉醇注射液(第五批集采价:68元/30mg)、注射用紫杉醇脂质体(228元/30mg)、注射用紫杉醇(白蛋白结合型)(1350元/100mg),以及经2024年国谈成功纳入谈判药目录的注射用紫杉醇聚合物胶束(376元/30mg)。相比于这些已有剂型,紫杉醇口服溶液的上市为患者提供了一种更为便捷的治疗选择。然而,在谈判与支付标准的制定过程中,如何将其剂型改良真正转化为患者获益,成为该药物能否成功进入医保的关键问题。作为化疗药物的创新形式,口服剂型的便利性是否能够显著提升患者的依从性并优化治疗结局,将直接影响其在医保准入谈判中的表现。
二、药物关键性临床试验的特点
在23个抗肿瘤药物中,有43.5%(10/23)药物通过附条件批准上市。此外,有超过半数(52.2%,12/23)抗肿瘤药物的关键性临床试验为单臂临床试验(Single Arm Trial,SAT)。在所有的SAT中,多数药物的临床试验分期为II期,有1个(8.3%,1/12)药物(匹妥布替尼)的临床试验分期为Ib期。在所有关键临床试验为随机对照试验(Randomized Controlled Trial,RCT)的药物中,临床试验分期均为III期。关键性临床试验分布情况见图3。
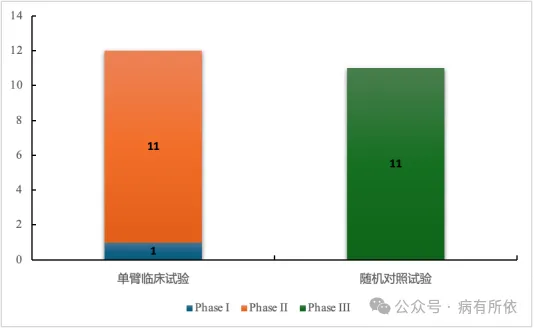
图3 关键性临床试验分布情况
获批的抗肿瘤药物关键临床结局成熟度较低,临床效果存在较大不确定性。在23个抗肿瘤药物中,仅有39.1%(9/23)同时观测到患者中位总生存期(Overall Survival,OS)和中位无进展生存期(Progression-free Survival,PFS);有65.2%(15/23)观测到患者中位PFS;有34.8% (8/23)患者中位OS和PFS均未观测到(详见图4)。关键性临床试验设计为SAT的药物中,所有的II期临床试验均以缓解率(包括客观缓解率、完全缓解率)作为主要临床结局,其中仅有33.3%(4/12)药物的关键性临床试验观测到了患者OS和中位PFS;有58.3%(7/12)观测到患者中位PFS,其余41.7%(5/12)未观测到成熟的OS和PFS。关键性临床试验设计为RCT的药物中,90.9%(10/11)药物的临床试验设置的主要临床结局为PFS,其中70%(7/10)药物的临床试验观测到相较于对照组有显著的PFS获益;仅有不到半数(45.5%,5/11)药物的临床试验观测到了患者中位OS,其中有且仅有60%(3/5)药物相较于对照组有显著的OS获益。药物临床试验中公布的PFS和OS数据是开展肿瘤药物经济学评价的重要参数来源,缺乏对照药品或关键结局不成熟势必会对高质量药物经济学证据的形成造成挑战,给药物报销决策带来不确定性。
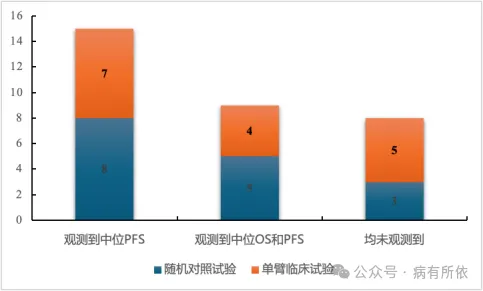
图4 药物临床试验中关键临床结局观测情况
三、国外卫生技术评估机构推荐意见
在23个抗肿瘤药物中,有9个(39.1%)药品可以查询到英国国家卫生与临床优化研究所(National Institute for Health and Care Excellence,NICE)或加拿大药物和卫生技术局(Canadian Agency for Drugs and Technologies in Health,CADTH)发布的推荐意见,所有的抗肿瘤药物均未被按获批适应症推荐报销。其中针对于莫妥珠单抗和芦比替定,HTA机构最终选择都是不推荐,其原因是这两个抗肿瘤药物获得上市批准的关键性临床试验均为单臂试验,HTA机构认为尽管制药厂商在递交的技术评估报告中采用间接比较的方法引入外部对照,但是会导致经济学评价结果存在较大的不确定性。此外,针对特定人群,临床试验严格的纳排标准导致人群证据缺乏和药品价格高导致ICER值过高,是HTA机构选择有条件推荐的主要原因。国外HTA机构推荐意见详见表1。(关注后私信“抗肿瘤药物”获取国外HTA机构推荐意见对应的理由)
表1 国外卫生技术评估机构推荐意见
药品名称适应症NICE推荐意见CADTH推荐意见
莫妥珠单抗淋巴瘤不推荐NA
替朗妥昔单抗淋巴瘤有条件推荐NA
芦比替定肺癌NA不推荐
他拉唑帕利前列腺癌NA停止评估
尼拉帕利阿比特龙前列腺癌NA有条件推荐
阿可替尼白血病有条件推荐有条件推荐
泊那替尼白血病有条件推荐有条件推荐
西达基奥仑赛多发性骨髓瘤停止评估有条件推荐
维恩妥尤单抗尿路上皮癌停止评估有条件推荐
四、创新抗肿瘤药物医保准入建议
药物经济性证据在药品医保目录准入及支付标准确定中发挥重要作用,故提交高质量的经济学评价证据既可以帮助制药厂商更精准定位谈判底价区间,又可以帮助医保决策者降低药品报销决策风险,提高药品谈判成功率并确定合理的支付标准,提升医疗资源配置和使用效率。药物经济学研究的效果参数通常来源于药物关键性临床试验,但随着药品监管部门附条件批准等快速审批程序的实施和医保目录调整工作的常规化进行,创新药物难以在上市后至提交医保目录准入申请的短时间内积累高质量临床效果证据。如何基于当前有限的临床证据生成相对高质量经济学评价证据,有以下几点建议:
1.基于单臂临床试验上市的药品,降低因外部对照引入的不确定性是关键。从国际HTA机构对于创新抗癌药物推荐意见及原因可知,引入外部对照导致的不确定性可能直接影响药物经济学评价结果的可信度以及医保准入的成功率。①选择优质间接比较方法:引入外部对照时采用匹配调整间接比较(MAIC)调整试验组与对照组之间的患者基线特征,降低因患者特征差异对药品效果评估的影响。②使用贝叶斯方法量化不确定性:利用贝叶斯统计框架,将外部对照的可靠性作为先验分布,结合单臂试验数据更新后验分布,量化不确定性。同时可以为决策者提供明确的不确定性范围,从而增强分析的透明度。③引入专家共识:对外部对照的选择、假设和模型进行专家评审,达成广泛共识。
2.开展上市后研究,填补现有临床效果证据空白。从第三部分国外HTA机构推荐意见梳理情况看,如果药品获批的适应症与关键性临床试验纳入患者人群存在差异,HTA机构通常会依据临床试验纳入的患者人群范围进行有条件的推荐。制药厂商应主动开展上市后研究(包括临床试验或真实世界研究),收集与获批适应症范围相符的患者数据,补充疗效和安全性证据,降低因部分患者人群效果证据缺失导致的经济学评价结果不确定性。
3.制药厂商构建与HTA机构的早期沟通机制。在药物尚未完全积累临床数据时,制药厂商可以通过与HTA机构的早期对话,获取对经济学评价模型设计的指导和反馈。这有助于确保药物经济学研究方法的科学性和透明性,并增加后续认可度。同时,与HTA机构的对接可以帮助为中国市场的药品提供合适的经济性分析框架。
 产业资讯
药智网 2026-03-24
40
产业资讯
药智网 2026-03-24
40
 产业资讯
氨基观察 2026-03-24
38
产业资讯
氨基观察 2026-03-24
38
 产业资讯
研发客 2026-03-24
55
产业资讯
研发客 2026-03-24
55
 微信公众号
微信公众号 热门资讯
热门资讯 热点标签
热点标签