

 产业资讯
产业资讯
 中国医药创新促进会
中国医药创新促进会  2025-02-15
2025-02-15
 1253
1253
引言
当全球生物医药产业驶入深水区,创新药“出海”已不仅是企业扩张的必选项,更是中国医药产业升级的战略支点。日本作为全球第三大医药市场,与中国药企在靶点开发、成本控制方面优势互补,成为中国创新药企出海的重要一站。
中国医药创新促进会(以下称“我会”)始终致力于深化中日医药产业合作,并在医药创新领域取得了一系列实质性成果。作为亚洲制药组织合作会议(Asia Partnership Conference of Pharmaceutical Associations,APAC)的成员,主导完成了多个具有重要意义、推动医药产业监管及创新发展的研究报告与学术论文;同时,作为日本制药工业协会(Japan Pharmaceutical Manufacturers Association,JPMA)的重要战略合作伙伴,定期举办中日医药企业交流活动,持续推动两国医药产业合作向纵深发展。2023年底,我会组织高级别医药产业代表团赴日考察交流,与日方研究机构及产业界开展了广泛深入的合作交流,为中国创新产品“出海”日本搭建赋能平台,助力中国创新成果惠及亚洲乃至全球患者。
为全面助力中国药监、生物技术公司、投资界深入了解日本药品监管法规、申报流程、沟通交流以及生产和检查等相关事宜,我会联合研发客、上海市生物医药科技产业促进中心以及泰格医药,共同开设“出海日本”专栏,特邀日本法规监管领域的资深专家发布专业性文章。撰稿人包括著名的药品开发及监管专家植村昭夫博士、东内祥浩先生和高野哲臣先生。同时还将对日本政府、学术界以及中日两国业内专家进行访谈,共同探讨开发及监管热门话题。
中国药促会中日医药合作交流
联系人:马明尧
电话:13520846026
邮箱:mamy@phirda.com
首篇文章今日正式发布。从本周开始至12月,专栏将保持每两周周五发布的频率,全年定期发行23期,解读日本的临床试验、市场及药品监管相关信息,力争化解创新药“出海”阻碍,推动创新成果惠及两国。
日本临床试验的历史(上)
2025年中文版第1期由系列A的高野负责,主题为“日本临床试验的历史(上篇)”,内容如下。本文也融入笔者的个人见解,并将探讨中日两国在药物临床试验起步阶段的历史差异以及由此导致的中日临床试验的不同特点。
01日本临床试验始于1960年代
日本的《药事法》(相当于中国的《药品管理法》DAL, Drug Administration Law)在第二次世界大战期间的1943年首次制定,引入了药品制造业的许可制度(前旧药事法)。随后,1948年制定了战后的新法规——旧药事法,政府许可事项大幅减少。为了建立以“国民皆保险”(所有国民都能加入保险并享受医疗服务的制度)为基础的健康保险制度,1960年对《药事法》进行了全面修订,日本的健康保险制度于1961年正式启动。(来源:维基百科)
当时获得批准生产的药品大部分是已在欧美销售的引进产品,根据1962年版的《药品制造指南》,临床部分只需提交日本2家临床研究中心60例以上的疗效判定数据即可获得批准。然而,1967年根据厚生省药务局长签发的通知《关于药品制造批准等的基本方针》,要求新药必须提交5家临床研究中心150例以上的临床研究数据。
随后,1980年4月修订的《药事法》实施,明文规定了必须确保药品等的质量、有效性和安全性。其中,首次将“临床试验”写入《药事法》,并制定了申办临床试验的标准、临床试验方案的申报等规定。
与此同时,各学会和关键意见领袖(KOL)主导制定了针对不同疾病的指南,如《降压药指南(1979年)》、《镇痛消炎药指南(1982年)》、《抗心绞痛药指南(1985年)》等依次出台。
日本关于药物临床试验实施的标准(旧GCP)于1985年12月公布草案,随后于1989年10月作为局长令公布,并于一年后的1990年10月正式实施。当时,临床试验受试者的口头知情也被允许。1996年5月,ICH-GCP达成一致并进入第4阶段,随后日本于1997年3月公布了GCP省令(新GCP),并于1998年4月全面实施,ICH-GCP得以落实,不再允许口头知情,而必须书面签署《知情同意书》。(来源:小清水敏昌《药史学杂志》49(1), 50-63(2014) 等)
02 1980年代以前日本临床试验均为国内本土试验
日本制药企业的创立时间分别为:武田制药1781年、山之内制药1923年和藤泽药品1894年(2005年合并为安斯泰来制药)、三共1899年和第一制药1915年(2007年合并为第一三共)、卫材1941年,这些企业均已有80年以上的历史。
1968年,以研发为导向的制药企业团体“日本制药工业协会(JPMA, Japan Pharmaceutical Manufacturers Association)”成立。响应1967年局长通知的要求,日本国内各制药企业开始对新药实施5家临床研究中心150例以上的临床试验,进入1970年代以后,日本进一步开始了以健康成人为对象的1期临床试验等,逐步推进自主研发的新药的国内开发和上市。
当时,直至ICH成立前的1980年代,日本尚未允许国际多中心临床试验(MRCT, Multi-Regional Clinical Trials),因此国产药的1期、2期、3期临床试验均为在日本国内进行的本土临床试验。 美国FDA自1970年代开始,便要求在注册临床试验中采用双盲试验设计,日本也在1980年左右开始引入双盲试验。
笔者于1986年加入安斯泰来制药(当时名为山之内制药)。当时的1期、2期、3期所有阶段的试验都需要包含日本人的临床数据,双盲试验已成为了标准。然而,当时的日本临床试验仍全部限于国内本土试验。
03 ICH成立与ICH-E6(GCP)和E5(种族因素)的制定以及日本桥接试验的开始
日本、美国和欧洲在药品上市前都需要药监部门评估和批准,因此各自建立了独立的法规体系。特别是在1960年代到1970年代,各国迅速完善了法规和指南,建立了新药质量、有效性和安全性的数据报告和评估体系。
然而,由于各地区对于注册申请的技术要求不同,制药企业需要重复进行大量试验以满足不同地区的法规要求。在此背景下,随着对药品开发成本不断增加的担忧,为了更快地向患者提供安全有效的新药,各地区药品审评审批标准的合理化和标准化显得尤为重要。
因此,1990年4月,日本、美国和欧洲的药品监管机构和药企团体的六方共同成立了ICH, International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use(国际人用药品注册技术要求协调委员会)。(来源:JPMA网站,ICH成立的背景)
ICH关于有效性的指南中,E6(GCP)于1996年6月10日E6(R1)达到第4阶段,如上所述日本于1997年3月公布了GCP省令(新GCP),并于1998年4月全面实施。作为增补于2016年11月9日E6(R2)达到第4阶段,日本于2019年7月公布,并于2020年1月实施。随后,作为全面修订于2025年1月6日E6(R3) Principles和Annex 1达到第4阶段,于2024年11月至2025年3月E6(R3) Annex 2公开征求意见。
此外,E5(种族因素)于1998年2月5日E5(R1)也达到第4阶段,日本在1998年8月以公告厚生省医药安全局长通知《关于国外实施的药品临床试验数据的处理意见》的形式,正式公布和实施了。
其他关于有效性的指南,如E1(长期给药试验)和E2A(临床试验中获得的安全性信息)于1994年10月、E3(总结报告)于1995年11月、E4(剂量-反应信息)于1994年3月、E7(老年人试验)于1993年6月、E8(一般指南)于1997年7月以及E9(统计原则)于1998年2月也都逐渐进展至第4阶段,均被各ICH成员国采纳。在1990年代,这些指南为“日本接受欧美的临床数据”或“日本、美国、欧洲进行国际多中心临床试验(MRCT)”奠定了基础。
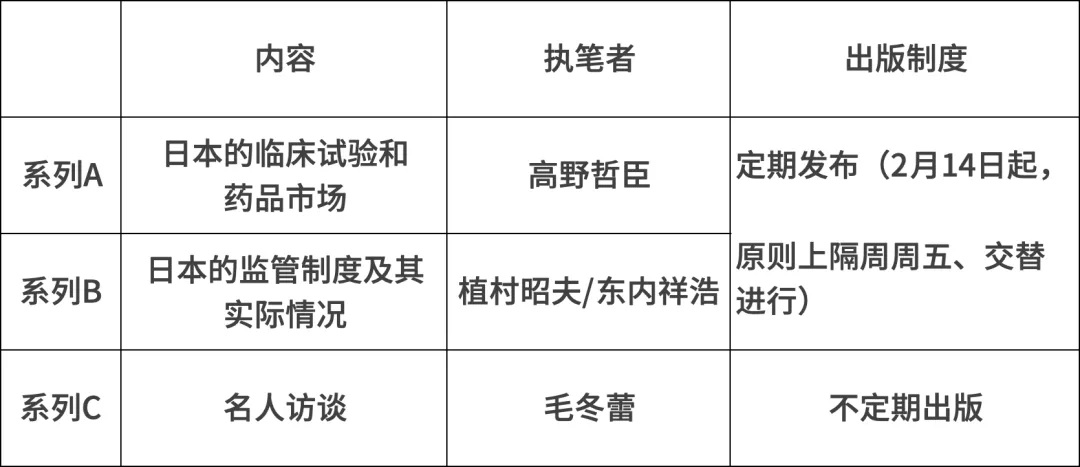
然而,实际上,在ICH E5(种族因素)于1998年公布和实施后,桥接策略在日本广泛实施,即利用欧美的1期、2期、3期临床数据在日本延迟进行2期桥接试验,导致日本在2005年之前参与的MRCT非常有限。(图1)
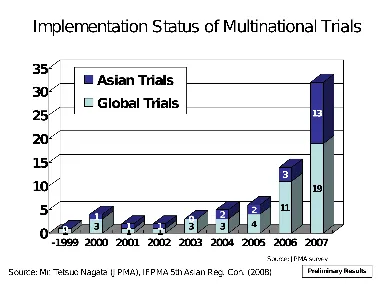
图1. JPMA成员企业在日本的MRCT数量(1998-2007)
(富永俊义, MHLW, 第6届韩日联合研讨会, 东京, 2008年4月11日)
这种日本临床试验国际化的滞后也反映在1997年GCP省令(新GCP)公布后,日本的临床试验数量减少到每年500项以下,直到2004年日本药品医疗器械综合机构(PMDA)成立,日本的临床试验数量一直未出现增长迹象。(图2)

图2. 日本的IND(临床试验申报)数量(1996-2010)
(森山佑辅, PMDA, 第2届中日医药交流会, 北京, 2011年3月29日)
04 日本从桥接试验到MRCT的战略转变
图3所示的是2012年5月在上海举行的第4届DIA中国年会上笔者的演讲幻灯片之一,展示了日本临床试验类型的变迁,顺序为A→B→C→D。
A.1997年日本采纳ICH-GCP之前,日本无法开展MRCT。另一方面,新药批准申请又需要提交1期、2期、3期数据。因此,在1997年以前,日本的临床试验均为境内本地试验。
B.1998年,ICH E5指南引入日本后,众多跨国制药企业在日本采用了桥接策略。这一策略导致在日本开展的许多临床开发项目出现了一个临床期(临床phase)的延迟现象。例如,日本的药代动力学(PK)试验往往在全球2期试验启动后才开始,日本的2期试验在全球3期试验开始后才开展。这种临床开发阶段的时间差,导致了日本的“药品上市延迟”。
C.2004年PMDA成立前后,PMDA和制药业界意识到,为了提升日本的公共卫生水平,并改善日本患者的治疗选择,必须消除“药品上市延迟”。由此,日本的药物开发战略迅速且彻底地由“桥接战略”转变为“MRCT战略”。然而,由于日美欧的临床开发项目启动时已经存在时间差,为了日本的尽快赶超,首先优先考虑日本参与全球3期试验。因此,当时也有跳过日本2期临床试验的临时对策。
D.PMDA鼓励制药企业在全球开发的早期阶段就将日本纳入。因此,从2010年左右开始,日本逐渐普及了与欧美同步的1期、2期、3期MRCT开发战略。
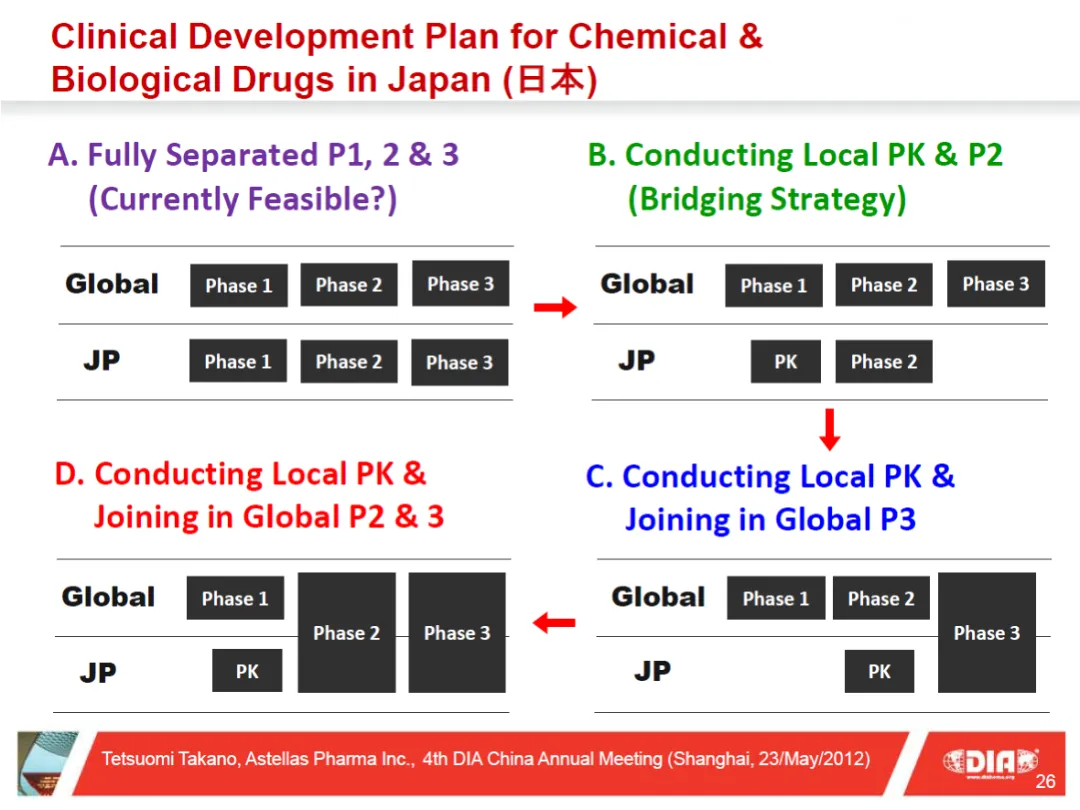
图3. 日本临床试验类型的变化历史
(高野哲臣, 安斯泰来制药, 第4届DIA中国年会, 上海, 2012年5月23日)
05 MRCT稳步增加的同时,药品错失和药品上市延迟再次出现
从2006-2007年开始,日本的MRCT崭露头角,并持续保持增长态势。截至2023年(令和5年),MRCT在日本临床试验中的占比已高达62.5%。(图4)
图4. 日本的MRCT IND(临床试验申报)数量(2007(H19)-2023(R05)
(松下俊介, MHLW, MID-NET Update 2024, 东京, 2024年11月28日)
另一方面,从日本的临床试验总数来看,自2022年以后,日本的临床试验数量大幅度减少,这印证了近年来日本的药品错失和药品上市延迟问题日益严重。(图5)
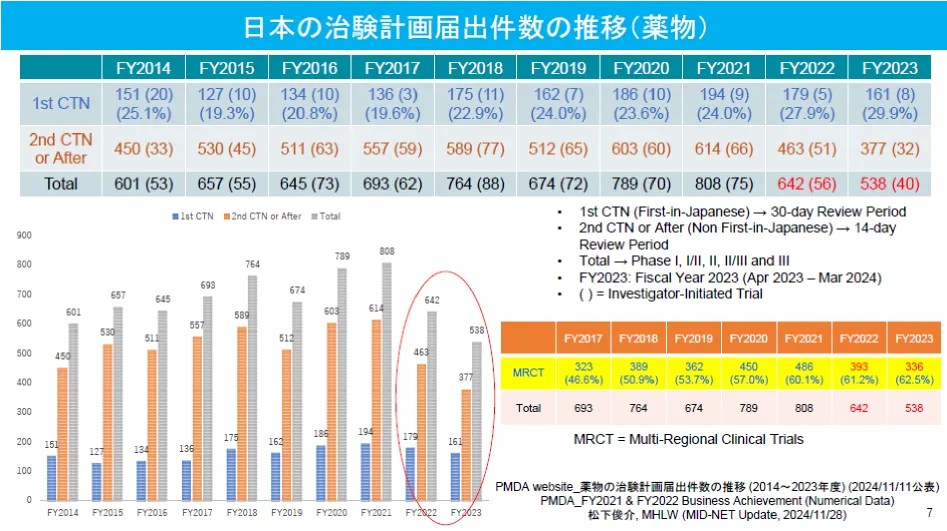
图5. 日本的IND(临床试验申报)和MRCT数量(2014-2023)
(高野哲臣, t2T Healthcare股份公司, 第31届红龙会研讨会, 东京, 2024年12月6日)
至此,《医药研发达人》中文版第1期“日本临床试验的历史(上)”就暂且搁笔。
关于2017年11月达到第4阶段并于2018年6月在日本实施的ICH E17(MRCT)的影响,以及2022年以后日本政府为应对药品错失和药品上市延迟所采取的措施对日本临床试验带来的变化,将在笔者后续撰写的《日本临床试验的历史(下)》中进行阐述。
 产业资讯
药智网 2026-03-24
15
产业资讯
药智网 2026-03-24
15
 产业资讯
氨基观察 2026-03-24
15
产业资讯
氨基观察 2026-03-24
15
 产业资讯
研发客 2026-03-24
30
产业资讯
研发客 2026-03-24
30
 微信公众号
微信公众号 热门资讯
热门资讯 热点标签
热点标签