

 产业资讯
产业资讯
 研发客
研发客  2025-03-29
2025-03-29
 5182
5182
2025年3月28日出版,医药研发达人中文版“出海日本”第5期(系列B《日本的监管制度及其实际情况》第二篇)
• 以药械法为核心,涵盖药品、医药部外品、化妆品、医疗器械及再生医疗等产品的监管,且法规会根据时代需求修订。
• 在日本,生产销售药品都受到药械法监管,需要获得监管部门许可或批准。涉及生产销售企业许可申请、生产销售注册申请、生产企业许可申请或外国生产企业认证申请、GMP符合性检查申请等四个关键步骤,每个步骤都有相应的申请资料和审查流程。
• 除药械法外,药品从开发到生产销售还需遵守GxP规范,包括GLP、GCP、GMP、QMS、GDP、GVP、GPSP等,其中部分是日本特有的规范,药事负责人需要充分理解。
本连载的系列B将介绍日本药品监管制度的基本信息。本期将谈一谈与日本药品批准申请相关的法规。
1. 日本药品监管相关法规
(1) 药事法、药械法
药事法于1960年颁布,当时颁布此法的目的是“规范药品、医药部外品、化妆品及医疗用具相关事项,确保其适当性”。此后,随着医疗领域中不断发生国际协调性、科技进步、企业行为多样化等变化,此法也逐步进行了适当的修订。2014年,医疗器械等被纳入其监管对象,此法也相应更名为《关于确保药品、医疗器械等的质量、有效性及安全性等的相关法律》,通称为药械法。
药械法的监管对象包括以下产品
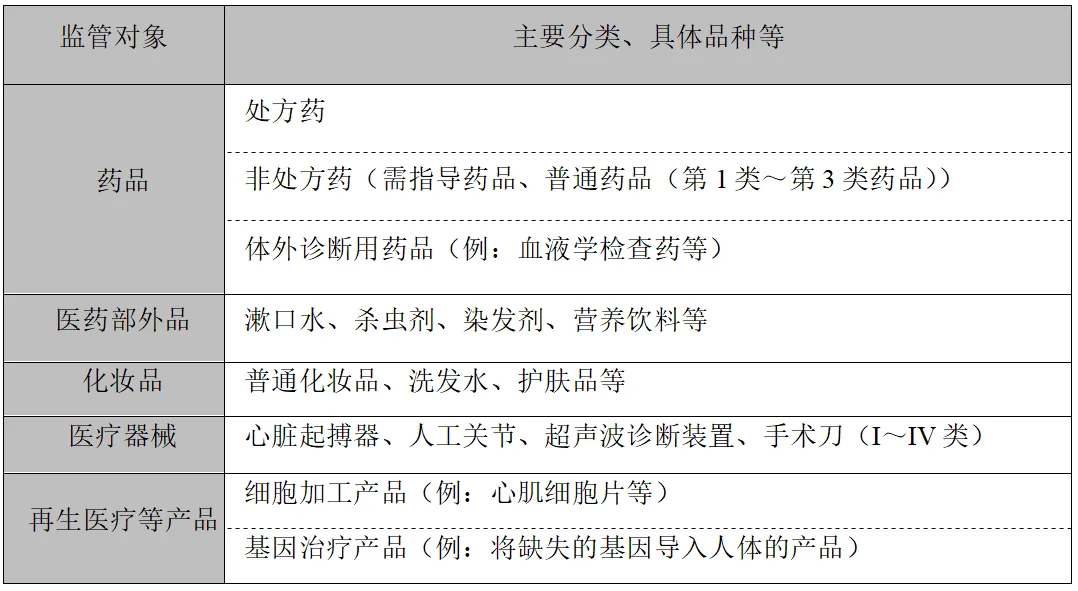
相反,保健品、健康食品、假睫毛、健康美容杂货、芳香剂等不在此法的监管对象范畴之内。
药械法是形成药品监管基础的重要法律,但根据时代需求,每隔几年就会进行一次修订,提前捕捉这些修订的变化并准备应对措施是制药企业的药事负责人的重要工作之一。
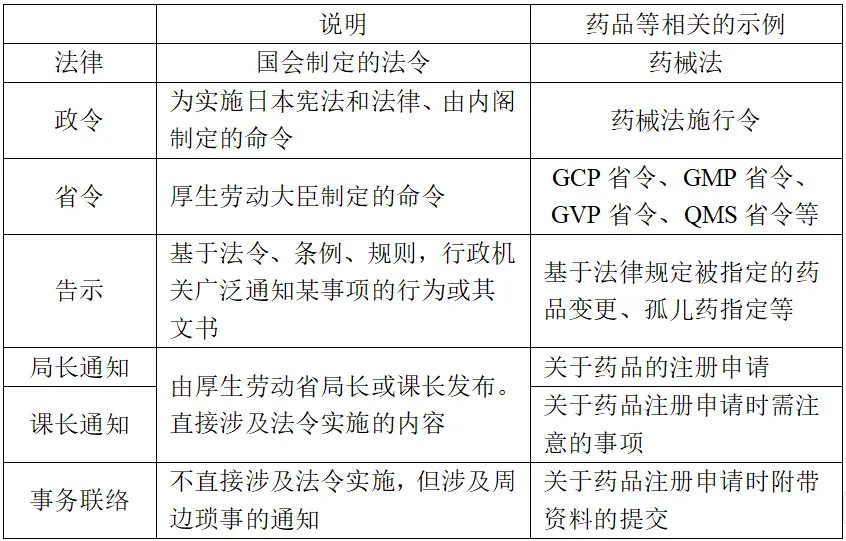
笔者精选信息3
日本的法规金字塔(层级型法规结构)
日本的法规金字塔如下。
2. 药械法中规定的药品生产销售程序
在日本生产销售药品受药械法监管,需获得监管部门(厚生劳动省(以下简称MHLW)及各都道府县)的许可或批准。生产销售应就以下四点进行申请并接受监管部门的审查。(A) 生产销售企业许可申请(B) 生产销售注册申请(C) 生产企业许可申请或外国生产企业认证申请(D) GMP符合性检查申请
(1) 企业代码
企业代码是管理药品、医疗器械等相关的企业许可及批准信息的管理系统中识别各企业的9位代码,在日本进行生产销售企业许可申请、外国生产企业认证申请等药事行为时是必需的,应在开展这些工作前取得。申请时,按照企业和生产厂分别填写企业代码登记表并提交给MHLW。
(2) 药品生产销售之前的程序
药品研发进展到准备注册申请阶段时,首先应取得企业代码,之后按以下流程接受对企业责任体系的审查((A)生产销售企业许可申请)。进一步接受对拟申报产品的生产方法及管理体系的审查(外国制造时:(C)外国生产企业认证申请)。所有准备完成后进行药品注册申请((B)生产销售注册申请)。此时,应就该产品的生产厂进行(D)GMP符合性检查申请。
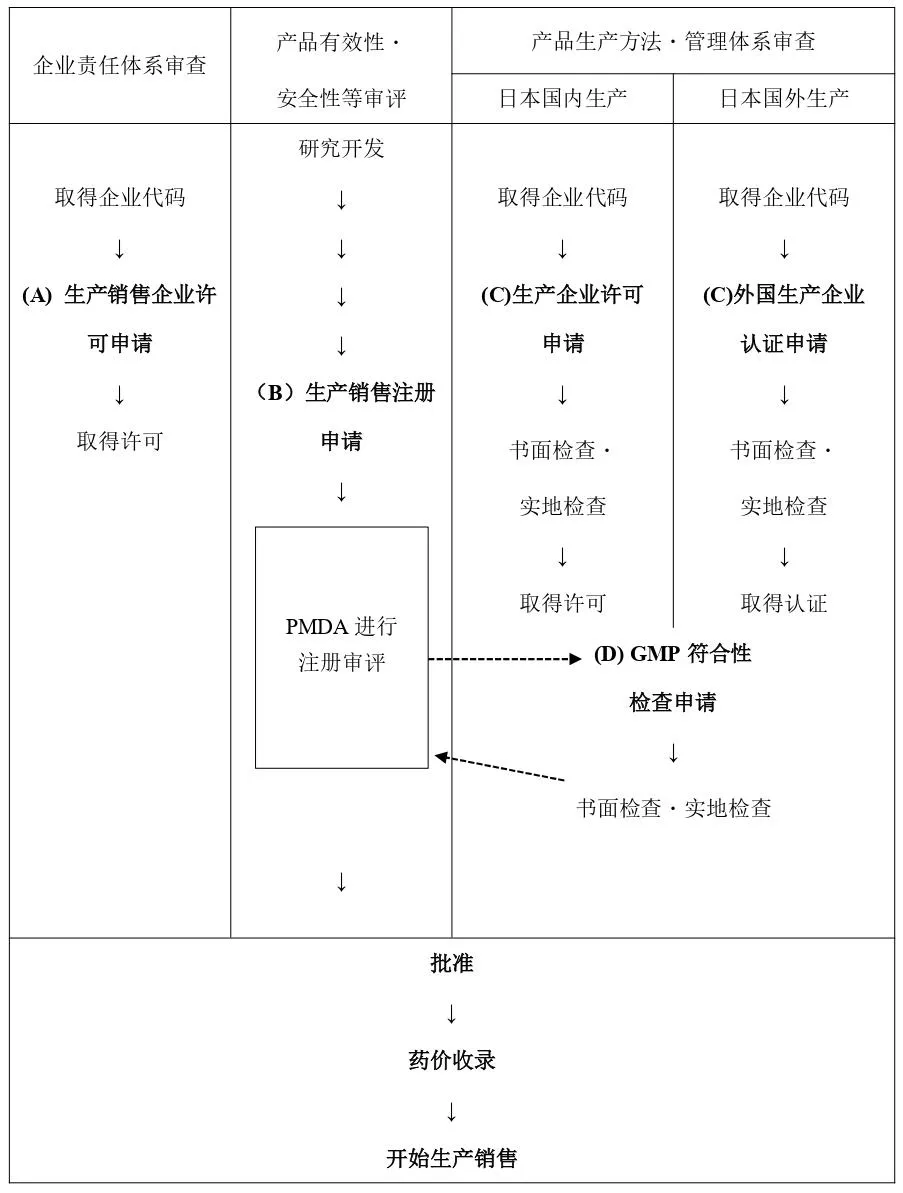
(A)至(D)的程序如下。
(A) 生产销售企业许可申请生产销售药品时,应向都道府县申报已具备承担产品市场最终责任、质量保证业务责任、安全管理业务责任的能力,并获得许可(即“企业许可”)。由申请人向各都道府县提交药品生产销售企业许可申请书,由知事在其权限下授予生产销售企业许可。生产销售企业许可需每5年更新一次。
・申请所需资料1. 登记事项证明书2. 负责药事相关业务公司的董事的(关于精神障碍和吸毒成瘾的)诊断书3. 生产销售总负责人的雇佣合同或证明雇佣关系的文件4. 生产销售总负责人的资格证明文件5. 组织架构图6. 质量管理体系文件7. 生产销售后的安全管理体系文件8. 配置图(同一场地内建筑物及建筑物内的该公司自用及其他公司的使用部分)9.事务所平面图10. 仓储设备图纸11. 事务所导览图(从最近车站到事务所的地图)
・补充说明:药品生产销售企业的三责为规范进行药品等的质量管理、安全管理,生产销售企业需设置“生产销售总负责人”、“质量保证负责人”及“安全管理负责人”(即“药事三责”)。
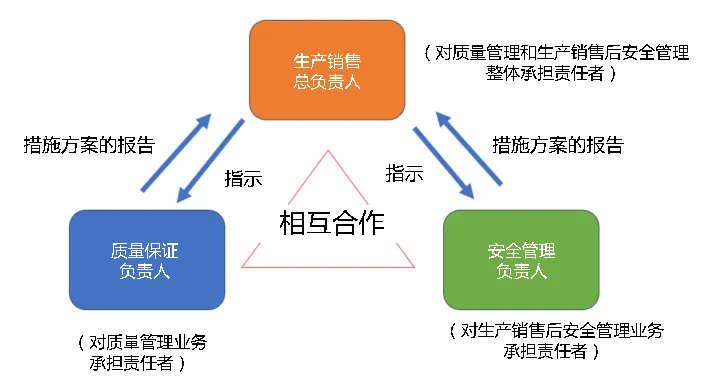
图1 质量管理、安全管理负责人
(根据https://www.mhlw.go.jp/stf/shingi2/0000168825.html改编)
(B) 生产销售注册申请相当于美国NDA或BLA,是日本的生产销售注册申请。需向MHLW申报药品本身在性能、安全性等方面无问题,并获得批准(生产销售批准)。一些已明确安全性的药品(已有批准标准的普通药品等)由都道府县知事批准。申请人向药品医疗器械综合机构(以下简称PMDA)或各都道府县提交药品生产销售注册申请书或外国生产药品的生产销售注册申请书,由厚生劳动大臣或各都道府县知事批准。
生产销售注册申请所需文件的详细信息将在今后的连载中介绍。
(C) 外国生产企业认证申请(产品生产方法・管理体系在海外时)日本国内药品生产企业需取得生产企业许可,委托外国生产企业生产药品时,需向MHLW申报该生产企业具备生产药品的能力,并获得认证。这与生产企业许可区分,称为外国生产企业认证。申请人向PMDA提交药品外国生产企业认证申请书,PMDA进行书面检查或实地检查,最终由厚生劳动大臣进行认证。外国生产企业认证需每5年更新一次。
药品的外国生产企业认证分为以下五种:1. 生物制剂等2. 放射性药品3. 无菌药品4. 普通5. 包装・标示・存储
・申请所需资料1. 认证申请书(如果由相关生产销售企业代办时,提供代办者名称・联系方式)2. 检查申请书3. 相关生产销售企业无法代办时,提供理由书及合同副本4. 业务分管表或组织架构图(附日文翻译)5. 生产厂负责人履历(附日文翻译)6. 生产品种一览表及生产工艺相关文件7. 生产厂结构设备相关文件8. 外国许可证等副本9. 翻译证明
(D) GMP符合性检查申请需向PMDA申报生产销售注册申请中涉及的生产厂符合“药品生产管理、质量管理标准”,并接受检查。申请人在提交生产销售注册申请后,向PMDA提交GMP符合性检查申请书。PMDA在生产销售注册申请的审评期间进行书面检查或实地检查,并将结果反映在药品注册审评中,本检查符合性是该药品批准的条件之一。
3. 药品GxP
除药械法外,药品从开发到生产销售需遵守的规范还有GxP。GxP是Good XXXX Practice的缩写,指药品研究、开发、试验、生产、流通、销售后的各专业业务的规范(省令)、指导原则等。GxP的例举如下。这些大多与欧美的国际规范协调,但其中存在与国际规范不同之处,也存在像GPSP这样的日本特有的规范,因此,药事负责人需对日本特有的规范有充分的理解。
GLP: Good Laboratory Practice(药品安全性相关非临床试验实施标准)
GCP: Good Clinical Practice(药品临床试验实施标准)
GMP: Good Manufacturing Practice(药品生产管理及质量管理标准)
QMS: Quality Management System(医疗器械及体外诊断用药品生产管理及质量管理标准)
GDP: Good Distribution Practice(日本国内药品运输・流通指导原则)
GVP: Good Vigilance Practice(药品生产销售后安全管理标准)
GPSP: Good Post-marketing Practice(药品生产销售后调查及试验实施标准)
 产业资讯
丁香园Insight数据库 2026-07-08
400
产业资讯
丁香园Insight数据库 2026-07-08
400
 产业资讯
招财小黄鸭 2026-07-08
349
产业资讯
招财小黄鸭 2026-07-08
349
 产业资讯
研发客 2026-07-08
477
产业资讯
研发客 2026-07-08
477
 热门资讯
热门资讯 微信公众号
微信公众号