

 产业资讯
产业资讯
 Insight数据库
Insight数据库  2025-04-30
2025-04-30
 4850
4850
2025 年 AACR 于 4 月 25 日至 30 日在美国芝加哥盛大召开。作为全球肿瘤领域学术盛会之一,本次大会上披露了诸多重磅临床试验的最新进展,如非小细胞肺癌、头颈部鳞状细胞癌等领域的临床研究。
Insight 数据库根据摘要内容梳理了 10 项值得关注的临床试验(不分先后),以洞察新药研发趋势。此外,Insight 数据库还在文末准备了 AACR 相关的资源大礼包,供参考下载。
默沙东:K 药作为头颈部鳞状细胞癌新辅助和辅助疗法的 III 期临床

截图来源:AACR 2025
早期研究显示,在局部头颈部鳞状细胞癌 (LA HNSCC) 患者中,SOC(手术+术后放疗±同步化疗)基础上添加新辅助和辅助免疫检查点抑制剂,能够取得良好的疗效。
KEYNOTE-689 是一项 III 期、随机、开放标签研究,在新诊断的 III/IVA 期、可切除、局部区域晚期头颈部鳞状细胞癌初治患者中,评估帕博利珠单抗术前给药作为新辅助治疗以及帕博利珠单抗联合标准放疗(联合或不联合顺铂)作为术后辅助治疗的疗效。研究的主要终点是无事件生存期(EFS)。
2024 年 10 月,默沙东首次宣布该试验达到其 EFS 主要终点。在首次中期分析中,接受帕博利珠单抗围手术期治疗方案的患者的 EFS 获得了统计学上显著且具有临床意义的改善,同时观察到 OS 改善趋势。
在关键次要终点上,该研究还显示,与单纯辅助放疗相比,帕博利珠单抗组患者的主要病理缓解 (MPR)也获得了统计学上显著改善,同时观察到 OS 改善趋势。
安全性方面,帕博利珠单抗的安全性与先前报告的研究结果一致,未发现新的安全性信号。
该试验具体的研究数据首次在 2025 年 AACR 大会上公布。363 例患者随机分配至帕博利珠单抗+SOC 组,351 例患者随机分配至 SOC 组。中位随访时间 38.3 个月,两组间的基线数据均衡,PD-L1 综合阳性评分 (CPS) ≥10 的患者群体中,帕博利珠单抗+SOC 组有 234 例患者,SOC 组有 231 例患者;CPS≥1 的患者群体中,帕博利珠单抗+SOC 组有 347 例患者,SOC 组有 335 例患者。
数据显示,在主要终点 EFS 方面,相较于 SOC 组,帕博利珠单抗+SOC 组所有患者的 EFS 为 51.8 个月(vs 30.4 个月,HR 0.73,95% CI 0.58-0.92,P = .00411),CPS ≥10 的帕博利珠单抗+SOC 组的中位 EFS 为 59.7 个月(vs 26.9 个月,HR 0.66,95% CI 0.49-0.88,P = .00217),CPS ≥1 的帕博利珠单抗+SOC 组的中位 EFS 为 59.7 个月(vs 29.6 个月,HR 0.70,95% CI 0.55-0.89,P = .00140)。
在次要终点 MPR 上,帕博利珠单抗+SOC 组和 SOC 组的差异具有统计学意义(9.3%,95% CI 6.7-12.8,P <.00001),CPS ≥10(13.7%,95% CI 9.7-18.7,P < .00001)和 CPS ≥1(9.8%,95% CI 7.0-13.3,P < .00001)的两个亚组中也具有显著性差异。另一次要终点 OS 数据将在进一步的随访中获得。
安全性方面,≥3 级 TRAE的发生率相似(帕博利珠单抗+SOC 组为 44.6%,SOC 组为 42.9%);TRAE 导致的死亡病例分别为 4 例和 1 例。43.2% 的帕博利珠单抗+SOC 组患者发生了免疫介导的不良反应,最常见的是甲状腺功能减退症。
总体而言,在 SOC 方案中添加帕博利珠单抗的新辅助和辅助疗法,显著改善了可切除局部头颈部鳞状细胞癌患者的 EFS 和 MPR 率,且与 CPS 无关。
值得一提的是,KEYNOTE-689 是首个证明抗 PD-1 疗法在早期头颈部鳞状细胞癌患者新辅助和辅助治疗中,EFS 具有统计学显著和临床意义改善的 III 期临床试验。
康方生物:派安普利单抗联合化疗一线治疗鼻咽癌的 III 期临床

截图来源:AACR 2025
Penpulimab(派安普利单抗)是目前唯一采用 IgG1 亚型并进行 Fc 段改造的新型差异化 PD-1 单抗,能够更有效增强免疫治疗疗效,且减少不良反应。
该药于 21 年 8 月首次在国内获批上市,用于治疗经典型霍奇金淋巴瘤,随后又先后在国内获批鳞状 NSCLC、鼻咽癌适应症。25 年 4 月 23 日,康方宣布该药基于国际多中心 III 期临床研究 AK105-304 和关键注册性研究 AK105-202 的积极数据在美国获批两项适应症,并且 AK105-304 研究数据将在 2025 年 AACR 大会上重磅发布。
AK105-304 是一项派安普利单抗联合化疗对比安慰剂联合化疗一线治疗复发或转移鼻咽癌的随机、双盲、III 期临床研究,在全球 46 个研究中心开展。主要疗效指标是 PFS,由盲法独立审查委员会根据 RECIST v1.1 标准进行评估。OS 是关键的次要终点。
291 例患者随机分配至派安普利单抗组(n=144)或安慰剂组(n=147)。截至 2024 年 4 月 29 日,中位随访时间为 19.1 个月。根据 BICR 评估:
派安普利单抗组和安慰剂组的中位 PFS 分别为 9.6 个月和 7.0 个月(HR=0.45,95% CI:0.33, 0.62,双侧 P < 0.0001)。
派安普利单抗组和安慰剂组确认的 ORR 分别为 68.1% vs. 63.9%,中位 DoR 分别为 9.8 个月 vs. 5.7 个月(HR=0.4, 95% CI: 0.27, 0.59)。
OS 尚未成熟,派安普利单抗组死亡 48 例,安慰剂组死亡 49 例(HR=0.94, 95% CI: 0.63, 1.40)。
安全性方面,派安普利单抗组和安慰剂组 ≥3 级 TRAE 发生率分别为 89.0% vs. 85.9%;SAE 发生率分别为 50.7% vs. 48.6%;irAE 发生率分别为 30.8% vs. 8.5%; ≥3 级 irAE 为 4.1% vs. 0。
翰森制药:阿美替尼作为 EGFR 突变 NSCLC 辅助疗法的 III 期临床

截图来源:AACR 2025
阿美替尼是国内首个上市的原研三代 EGFR TKI,可有效抑制致敏 EGFR 突变(ex19del/L858R)和耐药 T790M 突变。
此次 AACR 大会,翰森首次公布了阿美替尼作为 II-IIIB 期 EGFR 突变非小细胞肺癌(NSCLC)患者肿瘤完全切除后辅助疗法的 III 期 ARTS 研究结果(登记号:NCT04687241)。
ARTS 研究是一项随机、双盲、安慰剂对照的多中心 III 期临床试验,旨在评估阿美替尼对比安慰剂治疗 II-IIIB 期 EGFR 突变 NSCLC 患者(无论是否接受辅助化疗)的疗效和安全性。
研究共纳入 214 名中国患者,随机分配接受阿美替尼和安慰剂治疗。主要终点是 BICR 评估的无病生存期(DFS),次要终点包括研究者评估的 DFS、总生存期(OS)和安全性。BICR 评估的中位随访时间为 27.6 个月。
结果显示,阿美替尼组未达到 BICR 评估的 mDFS,而安慰剂组为19.4个月(HR 为 0.166,p<0.0001)。阿美替尼组 2 年 DFS 率为 88.2%,安慰剂组为 40.6%。研究者评估的 DFS 与 BICR 评估结果一致。数据截止时,OS 数据尚不成熟(奥莫替尼和安慰剂 OS 成熟度:2.8% vs. 3.8%)。
安全性方面,阿美替尼和安慰剂组导致剂量中断、剂量减少及停药的不良事件发生率分别为 12.3% vs. 17.8%、9.4% vs. 1.9% 和 0.9% vs. 0。未观察到新的安全风险。
总之,对于完全切除并接受辅助治疗的 II-IIIB 期 EGFRm NSCLC 患者,阿美替尼可显著改善患者的 DFS,且具有统计学和临床意义。
罗氏:TIGIT 单抗联合阿替利珠单抗治疗 NSCLC 的 III 期临床

截图来源:AACR 2025
Tiragolumab 是具有完整 Fc 区域的 TIGIT 单抗,也是全球首款进入 III 期临床的 TIGIT 单抗,而 Tecentriq(阿替利珠单抗)是靶向 PD-L1 的单抗。由于 TIGIT 通路与 PD-L1/PD-1 通路互补,Tiragolumab 和 Tecentriq 的联合方案被认为有可能克服免疫抑制并恢复免疫应答,进而提高疗效。
SKYSCRAPER-01 是一项全球性随机双盲 III 期研究,旨在 PD-L1 高表达、未经治疗的局部晚期不可切除或转移性 NSCLC 患者中评估 Tiragolumab 联合 Tecentriq(Tira + Atezo)与 Tecentriq 单药治疗的效果。患者按 1:1 的比例随机接受 Tira + Atezo 或安慰剂联合 Tecentriq(Pbo + Atezo)治疗,直至病情进展、临床获益丧失或出现不可接受的毒性。
2024 年 11 月 26 日,罗氏公布了 III 期 SKYSCRAPER-01 研究的最新进展,最终分析显示,该研究未达到总生存期这一主要终点。观察到的总体安全性与更长时间的随访结果一致,未发现新的安全性信号。
详细数据在本次 2025 年 AACR 大会上公布。该试验共有 534 名患者随机分组(Tira + Atezo,n=266;Pbo + Atezo,n=268)。中位随访 9.9 个月,经研究者评估,Tira + Atezo 组的中位 PFS 为 7.0 个月,Pbo + Atezo 组为 5.6 个月(HR 0.78;95% CI 0.63, 0.97;p=0.02)。在 OS 数据上,Tira + Atezo 组的中位 OS 为 23.1 个月,Pbo + Atezo 组为 16.9 个月(HR 0.87;95% CI 0.71, 1.08;p=0.22)。
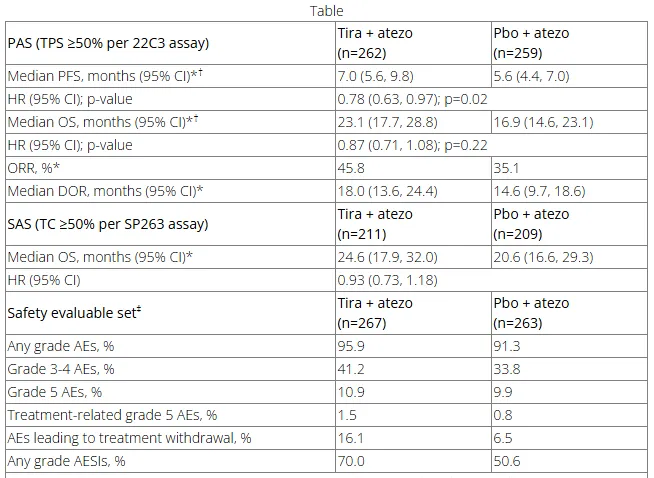
截图来源:AACR 2025
值得一提的是,在罗氏公布的 24 年财报中显示,Tiragolumab 被罗氏彻底放弃,包括 2 项 III 期临床、2 项 II 期临床、1 项 I 期联合治疗临床试验。
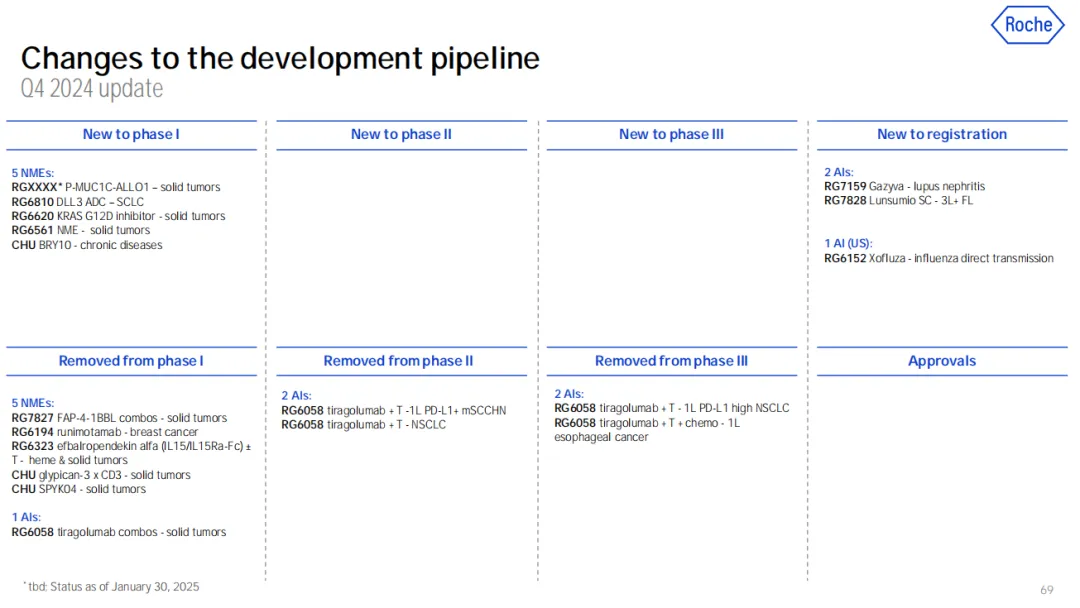
截图来源:罗氏 24 年财报
勃林格殷格翰:宗格替尼治疗伴有 HER2 基因异常实体瘤的 I 期临床
截图来源:AACR 2025
Zongertinib(宗格替尼)是勃林格殷格翰开发的一款不针对 EGFR 野生型的选择性 HER2 抑制剂。临床前研究显示,宗格替尼对所有主要的 HER2 突变(包括 HER2 YVMA 插入等位基因)均具有强效抑制的活性。
宗格替尼可选择性地共价结合 HER2 外显子 20 突变的受体酪氨酸激酶结构域。这种选择性使得 BI1810631 仅阻断 HER2 异常的下游信号传导,而不会影响野生型 EGFR(即正常 EGFR)信号通路,从而可避免其他泛-HER 抑制剂 TKI 常见的野生型 EGFR 相关的剂量限制性毒性。
2024 年 4 月,中国生物制药与勃林格殷格翰达成战略合作协议,双方将依托各自优势和资源,共同在中国内地研发和商业化勃林格殷格翰的肿瘤药物管线,其中就包括宗格替尼。
Beamion LUNG-1 是一项在 HER2 异常的晚期或转移性实体瘤患者中进行的宗格替尼单药治疗的开放性、Ia/Ib 期、剂量递增(含剂量确证和扩展)试验。研究的主要终点是 BICR 和研究者评估的 ORR。
Ib 期共有 5 个治疗队列,其中队列 1 纳入经治 HER2 酪氨酸激酶结构域(TKD)突变 NSCLC 患者,这些患者接受每日 120 mg 或 240 mg 宗格替尼治疗。经中期分析后,确定 120 mg 作为进一步评估的剂量。
此外,在队列 3 中,纳入经治 HER2 突变 NSCLC 患者,他们患有 TKD 或非 TKD 突变。在队列 5 中,纳入 HER2 TKD 突变 NSCLC 患者,这些患者预先接受过含铂化疗和 HER2 ADC 治疗。
2024 年 12 月,勃林格殷格翰在 2024 年 ESMO 亚洲大会公布了宗格替尼治疗 HER2 突变阳性晚期 NSCLC 经治患者的 Ib 期临床(BEAMION LUNG-1)队列 1 数据。
结果显示,宗格替尼(剂量为每日 120 毫克,单次服用,n=75 例患者)具有卓越疗效,客观缓解率(ORR)为 71%,疾病控制率(DCR)高达 93%。初步生存数据表明,宗格替尼缓解持久,6 个月无进展生存期(PFS)和缓解持续时间(DoR)比例分别为 69% 和 73%。
本次 AACR 大会,勃林格殷格翰公布了该试验更多的关键结果,包括队列 1 的中位 DOR 和中位 PFS,以及队列 3 和 5 的初步数据。
截至 2024 年 11 月 29 日,队列 1 中有 75 名患者,队列 3 中有 20 名非 TKD 突变患者,队列 5 中有 31 名患者,这些患者均接收 120 mg 宗格替尼治疗,并且每个队列分别有 33 例(44%)、 4 例(20%)和 13 例(42%)患者正在进行治疗。
在队列 1 中,经 BICR 评估的 ORR 为 71%,DCR 为 96%,中位 DoR 和 PFS 分别为 14.1 个月(6.9-NE)和 12.4 个月(8.2-NE)。
此外,存在脑转移的经治患者(n=27)中显示出颅内活性,ORR 达 41%,DCR 达 81%。
在队列 3 中,ORR 为 30%, DCR 为 65%,DoR 和 PFS 尚未成熟。
在队列 5 中,ORR 为 48%,DCR 为 97% ,DoR 和 PFS 分别为 5.3 个月(2.8-NE)和 6.8 个月(5.4-NE)。
安全性方面,在队列 1、3 和 5 中,分别有 97%/17%、80%/25% 和 77%/3% 的患者报告了药物相关不良事件(全部/G≥3),在所有队列中最常见的是腹泻。没有药物相关性间质性肺病(ILD)病例。
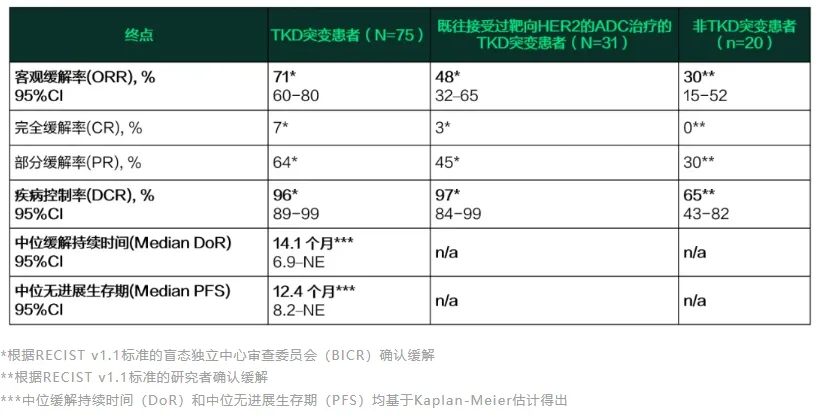
截图来源:企业官微
石药集团:SYS6010 治疗晚期实体瘤的 I 期临床

截图来源:AACR 2025
SYS6010 是由石药集团自主研发的重组人源化 EGFR ADC,含重组人源化抗 EGFR 单抗,该抗体通过连接子与拓扑异构酶 I 抑制剂 JS-1 结合。目前全球最高状态是 III 期临床。
临床前研究表明,SYS6010 能够剂量依赖性地抑制免疫缺陷小鼠中具有各种 EGFR 激活突变或野生型 EGFR 高表达的人类肿瘤的生长。特别是在含有针对第三代 EGFR-TKI 奥希替尼耐药的 EGFR 三重突变(Exon19Del、T790M 及 C797S)的人源化 NSCLC PDX 模型中,SYS6010 显示出很强的抗肿瘤效果。
本次 AACR 大会,石药集团首次披露该药的人体试验结果。这是一项多中心、开放标签的 I 期研究(CTR2023133),旨在评估 SYS6010 在晚期实体瘤患者中的安全性、耐受性、药代动力学特征和初步疗效。
该研究包括剂量递增、药代动力学扩展(第 1 部分)和剂量扩展(第 2 部分)两部分。第 1 部分采用 3+3 剂量递增设计,每 3 周静脉注射一次 6 个剂量水平的 SYS6010(0.6、1.8、3.6、4.8、5.6 和 6.4 mg/kg)并进行药代动力学扩展。第 2 部分是在第 1 部分确定的有效剂量下进行剂量扩展。
截至 2024 年 12 月 26 日,232 名患者接受了 ≥ 1 剂 SYS6010 治疗。最常见的癌症类型是 NSCLC(n=137)。既往抗癌药物治疗方案的中位数为 3 (1-11)。
174 例患者可进行疗效评估,ORR 和 DCR 分别为 32.8%(95% CI 25.85-40.27)和 86.2%(95% CI 80.18-90.96)。≥4.8 mg/kg 剂量组 ORR 和 DCR 分别为 39.1%(45/115)和 78.3%(90/115)。
在各亚组人群中,尤其是对 EGFR TKI 耐药或 EGFR 野生型的 nsq-NSCLC 患者,SYS6010 具有良好的疗效:
EGFR 突变非鳞状 NSCLC(nsq-NSCLC)≥ 4.8 mg/kg 组的 ORR 和 DCR 分别为 50%(27/52)和 92.3%(48/52)。
在既往 EGFR TKI 治疗失败的 EGFR 突变 NSCLC 的受试者中,ORR 和 DCR 分别为 90%(9/10) 和 100%(10/10)。
对于既往 EGFR TKI 联合铂类化疗失败的患者,ORR 和 DCR 分别为 41.5%(17/41) 和 90.2%(37/41)。
在免疫治疗和化疗均失败的 EGFR 野生型 nsq-NSCLC 患者中,≥ 4.8 mg/kg 剂量组的 ORR 和 DCR 分别为 50%(3/6)和 83.3%(5/6)。
在安全性方面,6.4 mg/kg 剂量组中发生一例限制性毒性(4 级血小板减少症),未达到 MTD。91.8% 患者发生治疗相关不良事件(TRAE),其中 ≥ 3 级 TRAE 占 47%。最常见 TRAEs 为白细胞减少、贫血、恶心、血小板减少 、中性粒细胞减少、食欲下降、乏力、呕吐、皮疹和脱发。
翰森制药:阿美替尼联合化疗一线治疗 EGFR 敏感突变 NSCLC 的 III 期临床

截图来源:AACR 2025
这是一项随机、对照、开放、多中心、III 期研究(NCT04923906),旨在评估阿美替尼联合化疗对比阿美替尼单药一线治疗 EGFR 敏感突变局部晚期或转移性 NSCLC 的疗效与安全性。
2024 年 10 月,翰森首次公开宣布该临床试验达到 PFS 的主要终点。阿美替尼联合化疗的患者在疾病进展或死亡的风险方面,风险降低超过 50%,具有统计学显著性,中位无进展生存期延长至超过 2 年。
此次 AACR 大会,翰森公布该试验的详细数据。截至 2024 年 6 月 18 日,共有 624 例患者入组(阿美替尼联合化疗组 310 例,阿美替尼单药治疗组 314 例),按 EGFR 突变类型(Ex19del vs. L858R)和基线 CNS 转移状态分层。
中位随访时间为 23.4 个月,联合化疗组中位 PFS 为 28.9 个月(95%CI 26.3-NA),单药组为 18.9 个月(95%CI 17.8-21.1),风险比为 0.471(95%CI 0.371-0.598;P<0.0001),所有预设亚组显示一致获益。OS 数据尚不成熟,HR 为 0.442(95%CI 0.308-0.636;P<0.0001)。
在安全性方面,联合化疗组和单药组 ≥3 级不良事件发生率分别为 75.7% vs 23.7%;导致阿美替尼停药的不良事件发生率分别为 3.0% vs. 1.3%。培美曲塞中位治疗周期为 20.0 个,88.8% 的患者完成 4-6 个周期的铂类化疗。联合方案的安全性特征与单药治疗已知的安全性特征一致。
默沙东:奥拉帕尼联合 K 药治疗 HRRm/HRD 晚期癌症的 II 期临床

截图来源:AACR 2025
早期数据表明,PARP 抑制剂/抗 PD-(L)1 抗体在同源重组修复突变 (HRRm) 或同源重组缺陷 (HRD) 癌症中具有协同作用。
KEYLYNK-007 (NCT04123366) 是一项不限癌种的 II 期临床试验,评估了 PARP 抑制剂奥拉帕尼联合帕博利珠单抗治疗 HRRm/HRD 晚期癌症的疗效。
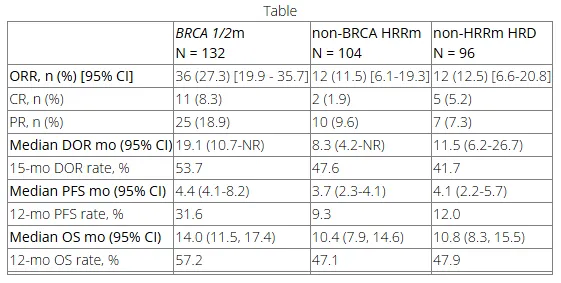
这项 II 期临床试验将患有 HRRm 或 HRD 阳性的晚期实体瘤受试者分为 3 组,包括 BRCAm(乳腺癌和卵巢癌除外)、非 BRCA HRRm 和非 HRRm HRD。主要终点是 ORR。次要终点是缓解持续时间和 PFS、OS 和安全性。
截至 2024 年 5 月 16 日,共入组 332 名患者,数据显示,BRCAm 组的中位随访时间为 13.4 个月,ORR 为 27%;非 BRCA HRRm 组的中位随访时间为 10.4 个月,ORR 为 12%;非 HRRm HRD 组的中位随访时间为 10.8 个月,ORR 为 12%,
在 PARP 抑制剂获批适应症之外,确认获得缓解的适应症包括晚期乳腺癌、卵巢癌、胰腺癌、泌尿上皮癌、肾癌、胆道癌、宫颈癌、食管鳞癌、结直肠癌、胃食管结合部腺癌、十二指肠癌、小肠癌、甲状腺癌、唾液腺癌、头颈部鳞状细胞癌、NSCLC、小细胞肺癌、平滑肌肉瘤及其他肉瘤,HRRm 前列腺癌也有多例缓解。
在安全性方面,30% 的患者发生了 ≥3 级药物相关不良事件,0.3% 的患者死于感染性休克。
总体而言,在这项不限癌种的试验中,奥拉帕尼联合帕博利珠单抗在 HRRm 和HRD 阳性(尤其是 BRCAm)晚期癌症中表现出良好的持久抗肿瘤活性和可控的安全性,超出了目前已获批的适应症。
截图来源:AACR 2025
罗氏:RO7589831 治疗 MSI 和/或 dMMR 晚期实体瘤的 I 期临床

截图来源:AACR 2025
WRN 是一种对 DNA 修复和基因组稳定性至关重要的酶,是微卫星不稳定性(MSI)癌症有潜力的合成致死靶点。40%-70% 的 MSI/错配修复缺陷(dMMR)实体瘤患者对免疫检查点抑制剂没有反应或产生耐药性。
RO7589831 是由罗氏和 Vividion Therapeutics 合作研发的一种口服新型首创共价不可逆 WRN 抑制剂。
2024 年 AACR 大会上,Vividion Therapeutics 披露了该药的结构和初始临床前药理学数据。该药在多种 MSI 结直肠癌细胞系和患者来源的异种移植模型(包括接受免疫检查点疗法治疗后病情进展的患者模型)中均表现出显著的肿瘤消退。
本次公布的是一项开放标签、多中心、I 期临床研究 (NCT06004245) ,评估了 RO7589831 对 MSI 和/或 dMMR 晚期实体瘤患者的安全性、药代动力学 、药效学和初步抗肿瘤活性。主要目的是确定最大耐受剂量 (MTD) 和/或推荐的 II 期剂量 (RP2D)。
截至 2024 年 11 月 25 日,共 44 例患者(22 名 CRC 和 22 名非 CRC)被纳入 6 个剂量组(从 150 mg 到 2000 mg)。这些患者既往治疗线数中位数为 3,89% 接受过免疫检查点抑制剂治疗。
在 32 名疗效可评估的 MSI 患者中,4 名患者获得部分缓解(PR)(2 例已确认),持续缓解时间长达 9.5 个月以上,DCR 为68.8%(95%CI 51.13, 86.37),20 例患者达到疾病稳定(SD)。
在安全性方面,最常见的治疗中出现的 AE 是恶心、腹泻和呕吐。3 级 (G) 治疗相关 AE (TRAE) 包括恶心和 AST/ALT 升高以及疲劳和贫血。未观察到 G4 或更高级别的 TRAE。
君实生物:JS107 治疗晚期实体瘤的 I 期临床

截图来源:AACR 2025
JS107 是一款 Claudin18.2 ADC,可以与肿瘤细胞表面的 Claudin18.2 结合,通过内吞作用进入肿瘤细胞内,释放小分子毒素 MMAE,对肿瘤细胞产生强大的杀伤力。 并且由于 MMAE 的细胞通透性,JS107 能够通过旁观者效应介导对其它肿瘤细胞的无差别杀伤,从而提高疗效并抑制肿瘤复发。
这是一项开放标签、单药或联合治疗、剂量递增和剂量扩展的首次人体 I 期临床研究(NCT05502393)。主要研究终点是安全性,次要终点包括疗效和药代动力学特征。
单药研究的剂量递增阶段纳入标准治疗失败的晚期实体瘤患者,接受 JS107(0.15-3.5 mg/kg, Q3W)治疗,剂量扩展阶段纳入 CLDN18.2 阳性患者,接受 JS107(2.0 或 3.0 mg/kg, Q3W)治疗。
联合治疗研究纳入既往未接受过治疗的 CLDN18.2 阳性、HER2 阴性的晚期胃/胃食管结合部癌(GC/GEJ)患者,接受 JS107(剂量递增阶段:2.0-3.0 mg/kg, Q3W;剂量扩展阶段:2.0 mg/kg, Q3W)联合特瑞普利单抗(240 mg, Q3W)和 XELOX(卡培他滨+奥沙利铂)治疗。
截至 2025 年 1 月 7 日,单药研究共入组 63 例患者(剂量递增阶段 22 例,剂量扩展阶段 41 例),联合治疗研究共入组 27 例患者(剂量递增阶段 9 例,剂量扩展阶段 18 例)。结果显示:
JS107 单药治疗尚未达到最大耐受剂量(MTD),联合治疗 MTD 为 2.5 mg/kg;
接受 JS107 2.0-3.0 mg/kg 单药治疗的 CLDN18.2 高表达(定义为 ≥20% 肿瘤细胞中 CLDN18.2 染色强度≥2+) GC/GEJ 患者中,ORR 为 34.8% (8/23),中位 PFS 为 4.11 个月;
接受联合治疗的 CLDN18.2 高表达的 GC/GEJ 患者接受(14 例疗效可评估),ORR 达 78.6%(11/14);
单药和联合治疗研究中,均观察到 CLDN18.2 表达水平与疗效呈正相关。
药代动力学分析显示,JS107 治疗剂量 0.15-3.5 mg/kg 之间,ADC 和总抗体暴露量呈剂量依赖性。
在安全性方面,单药和联合治疗的 ≥3 级 TRAE 发生率分别为 47.6% 和 40.7%,其中最常见的分别为中性粒细胞减少 (22.2%) 和血小板减少 (18.5%)。
总体而言,在 CLDN18.2 高表达晚期 GC/GEJ 患者中,JS107 单药或联合特瑞普利单抗+XELOX 治疗均显示出良好的疗效,且安全性可控。Insight 数据库显示,在胃癌 CLDN18.2 ADC 领域,此前披露的所有临床研究数据均建立在单药治疗的基础上,而君实的此项研究是首个证明 CLDN18.2 ADC 联合疗法在胃癌患者具有临床获益。
 产业资讯
佰傲谷BioValley 2026-05-14
485
产业资讯
佰傲谷BioValley 2026-05-14
485
 产业资讯
丁香园Insight数据库 2026-05-14
636
产业资讯
丁香园Insight数据库 2026-05-14
636
 产业资讯
药智网 2026-05-14
528
产业资讯
药智网 2026-05-14
528
 热门资讯
热门资讯 微信公众号
微信公众号