

 产业资讯
产业资讯
 药智网
药智网  2025-07-15
2025-07-15
 5763
5763
近期,NMPA发布《优化创新药临床试验审评审批有关事项(征求意见稿)》,提出将创新药临床试验默示许可的审评时限由60日压缩至30日。制度加码、监管提速,一场围绕创新药审批逻辑的“结构性变革”正在悄然发生。
01
从60天到30天,这场改革改了什么?
“30天”不是简单的时间砍半,而是新药临床试验申请审评制度的一次系统性升级。
6月13日,为落实《国务院办公厅关于全面深化药品医疗器械监管改革促进医药产业高质量发展的意见》(国办发〔2024〕53号)有关要求,支持创新药研发,国家药监局在开展优化创新药临床试验审评审批试点工作经验基础上,组织起草了《关于优化创新药临床试验审评审批有关事项的公告(征求意见稿)》。
此次政策征求意见稿围绕“压时增效”核心目标,聚焦1类化学药、1类生物制品与创新中药,提出多项实质性调整:
将创新药临床试验申请(IND)的审评期限从60天压缩至30天,重点支持儿童药、罕见病、新机制产品和全球同步研发品种。明确“审评启动时间点”为资料提交日,变原先的“受理日倒计时”为“提交即倒计时”,为企业节省流程等待时间。
该政策背后反映出监管层对“提速同时不降质”的制度信心,监管从传统的逐项审批向“风险管理+数据审查”机制演进。
表1 60天 vs 30天审批对比
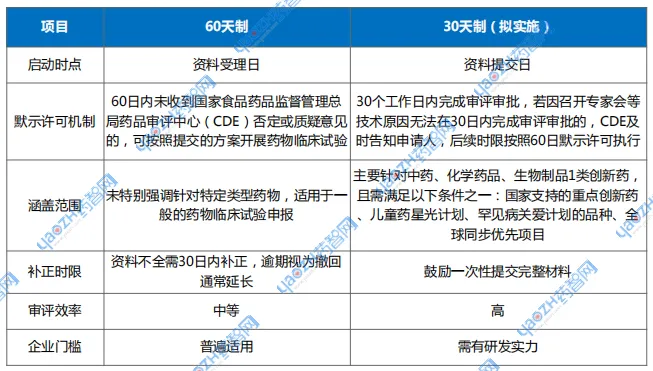
资料来源:公开资料,药智咨询整理
中国IND审评时限的压缩,并非一蹴而就,而是沿着“改革—试点—制度化”的路径逐步推进。
早在2018年,国家药监局正式引入60日默示许可制度,开启我国从审批制向风险导向审评机制的首次转型(下表2)。
此后,根据药智数据企业版——药品注册与受理数据库的智能统计,IND申请的实际总受理时长也在逐年降低:由初期约130天,逐步缩短至当前的70天左右(下图1)。
为进一步提速,2024年7月,国家药监局发布《优化创新药临床试验审评审批试点工作方案》,提出在部分区域与机构试点将IND审评压缩至30个工作日以内,以切实“缩短药物临床试验启动时间”。试点计划包括:2024年8月底前完成机构遴选,2025年1月启动中期评估,2025年7月完成总结并提出机制固化建议。
在此基础上,2025年6月,国家药监局发布《优化创新药临床试验审评审批有关事项(征求意见稿)》,提出将“30日通道”从局部试点走向全国推广。
从60天到30天,从点上试点到制度成型,中国IND审评制度已形成“政策设计—机制验证—制度落地”的完整闭环,全面迈入以“效率提升与科学监管”为导向的新阶段。
表2 中国临床试验申报审评审批时长相关政策变化
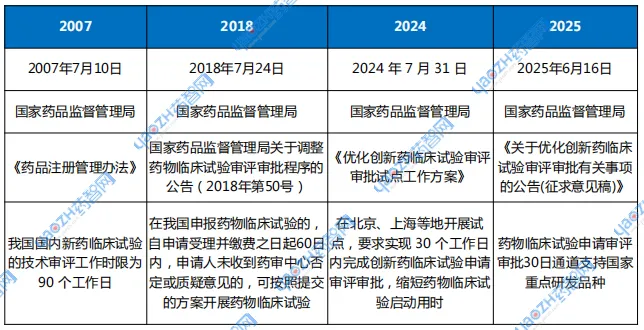
资料来源:NMPA官网,药智咨询整理
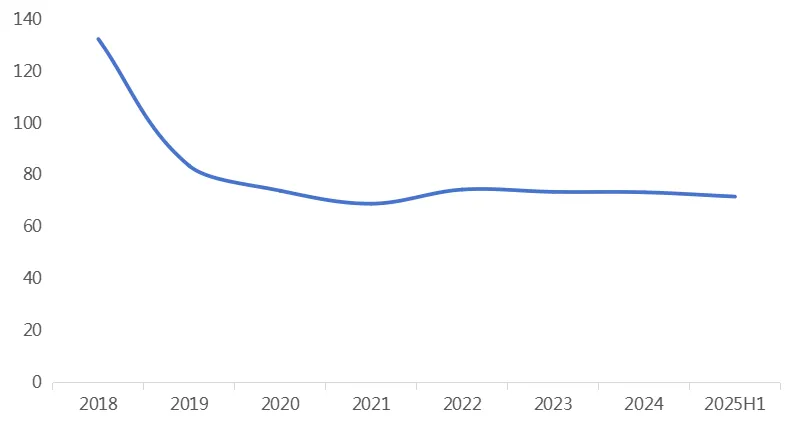
图1 2018-2025年上半年,创新药IND总受理时长
资料来源:药智数据(审评时长智能计算),药智咨询整理
值得注意的是,意见稿中还鼓励申报企业同步准备伦理审查、药学研究和临床试验资料,推动各环节协同加速。政策设计更关注申报质量,要求企业在研发早期即遵循“上市导向”策略。
从管得快,到用得上,中国创新药加速不仅在审评端,更在支付侧补上“最后一公里”。与此同时,医保支付侧也在主动跟进创新药的制度保障。
2025年7月,国家医保局正式发布《支持创新药高质量发展的若干措施》,明确提出通过多元支付路径(如医保准入、医保目录动态调整、商业保险联动)支持真正具备临床价值的创新药上市与使用。与NMPA推进的“临床快审批”形成合力,中国创新药正在迎来“研发—审批—支付”三位一体政策闭环的新格局。
02
头部玩家的“加速度游戏”
30天通道,不是普惠式快车,而是系统能力的综合考试。
于中国药企而言,30日通道的设立无疑是一次重大制度红利。那些拥有成熟研发能力、强大CMC储备和快速响应注册策略的企业,将率先受益。例如百济神州、上海翰森、正大天晴、江苏恒瑞等企业,在全球同步、早期临床、肿瘤赛道上已有深厚积累,将成为第一批“跑在前”的获益者。
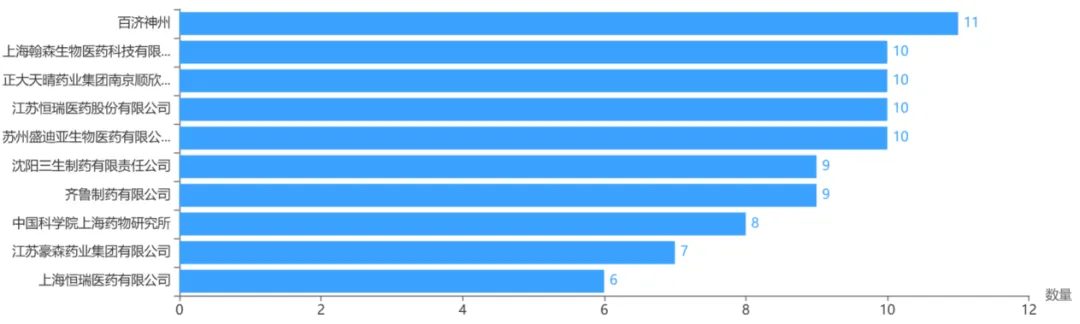
图2 2025年上半年中国1类新药临床试验申请药品数量TOP10企业
图片来源:药智数据
与此同时,资本市场也将重新定价“早期+快通道”资产。
以往早期项目因风险大、审批周期长而估值较低,如今审批窗口大幅提速,给了Pre-IND及IND阶段项目更高溢价空间。尤其对于拟开展全球同步临床的产品,制度明确支持将加速吸引海外资本与合作方参与。
但政策红利的另一面,是中小型药企所面临的现实挑战。
尽管时间缩短,但并不意味着审评标准降低。30天的倒计时要求企业在项目立项初期就考虑临床定位、数据完整性、伦理审核与CMC保障,缺乏系统整合能力的企业可能反而”抢跑翻车”。
03
中美IND审批对比:向创新药强国看齐
回顾中美两国的临床审批制度,不难看出中国此轮改革的方向和目标。
在美国,FDA自1997年起即实行IND 30天审评制,强调申报资料完整性、临床试验设计合理性及申办方资质。更关键的是,美国高度依赖注册前沟通(Pre-IND Meeting),通过早期沟通避免后期反复,有效提升审评效率。FDA审评强调“科学导向+风险评估”模型,已逐步形成制度闭环。
相比之下,中国2018年引入60天默示许可是重大转型,此次拟推行30天审评,是向FDA靠拢的“第二跳”。
表3 美国vs中国IND评审对比
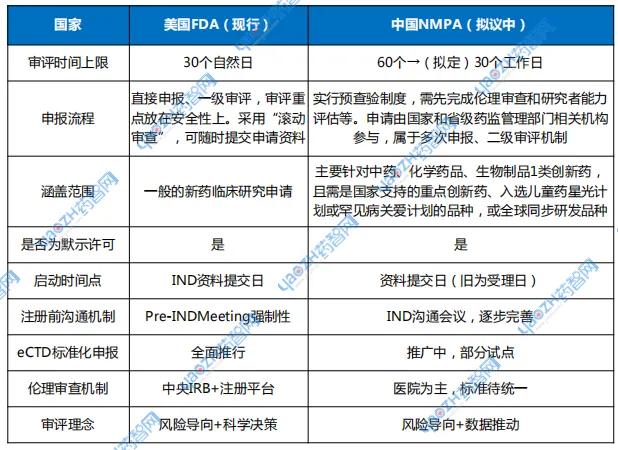
资料来源:公开数据,药智咨询整理
但中美在配套机制方面仍有差距,例如伦理审查独立性不足、申报资料标准不统一、监管与行业沟通机制尚不成熟。
此次NMPA缩短审评周期,不仅是制度效率上的追赶,更是理念上的融合:中国药品监管开始拥抱“科学评审、风险管理、沟通优先”的国际主流模式。这为中国创新药参与全球竞争打下基础,也为企业出海、BD合作创造更高制度兼容度。
04
结语
30日审批不是口号,而是中国新药制度从效率走向质量、从合规走向全球化的又一信号。它给予有准备者以速度红利,也为制度进化按下快进键。新一轮的临床竞速已经开始,只有那些真正具备系统能力与战略前瞻的企业,才能真正驶上这条“快车道”。
 产业资讯
瞪羚社 2026-06-18
381
产业资讯
瞪羚社 2026-06-18
381
 产业资讯
深蓝观 2026-06-18
397
产业资讯
深蓝观 2026-06-18
397
 产业资讯
研发客 2026-06-18
440
产业资讯
研发客 2026-06-18
440
 热门资讯
热门资讯 微信公众号
微信公众号