

 研发追踪
研发追踪
 生物药大时代
生物药大时代  2025-07-22
2025-07-22
 2512
2512
在上一周,美国FDA先后驳回罗氏(Roche)旗下基因泰克(Genentech)的CD20/CD3双特异性抗体Columvi,以及葛兰素史克(GSK)的BCMA靶向抗体偶联药物(ADC)Blenrep的上市申请。两款药物均针对血液肿瘤领域未满足的临床需求,且此前被寄予厚望。
Blenrep:未达到OS关键次要终点,临床入组人群代表性欠缺,安全性存疑
2025年7月17日,葛兰素史克(GSK)宣布FDA肿瘤药物咨询委员会(ODAC)以5:3的投票结果反对其BCMA ADC新药Blenrep用于复发/难治性多发性骨髓瘤(R/R MM)的二线联合治疗,理由是认为其风险大于收益。Blenrep的PDUFA日期为7月23日。

Blenrep是GSK公司的一款靶向B细胞成熟抗原(BCMA)的ADC。由人源化的BCMA单抗通过马来酰亚胺(mc)连接子,与微管蛋白抑制剂单甲基澳瑞司他汀F(MMAF)偶联而成。早在2020年,Blenrep曾通过加速审批在美国上市,用于治疗接受过至少两线疗法的多发性骨髓瘤患者。然而,由于在一项关键的确认性试验中未能证明其在无进展生存期(PFS)方面优于标准治疗方案,Blenrep于近三年前被FDA要求撤市。
Blenrep此次上市申请基于两项全球III期临床试验DREAMM-7和DREAMM-8的数据。DREAMM-7显示,Blenrep联合硼替佐米和地塞米松(BVd)相比标准疗法,将中位无进展生存期(PFS)从13.4个月延长至36.6个月,死亡风险降低42%,三年生存率达74%。然而,DREAMM-8试验中,Blenrep单药治疗组的中位PFS未达终点(12.7个月 vs 对照组12.7个月),且总生存期(OS)数据未显著改善。ODAC指出,DREAMM-8未达到预设的OS关键次要终点,且两项试验入组人群中美国患者入组率低(每项研究中低于 5%),质疑结果对美国人群的适用性。
此外,FDA指出在葛兰素史克两项3期临床试验中观察到的“眼部毒性发生率很高”。具体而言,两项研究中的大多数患者都出现了角膜病变和视力下降,并且治疗还“与严重的眼部毒性有关”,例如角膜溃疡。FDA还指出,在DREAMM-7和DREAMM-8两项试验中,Blenrep的剂量设计存在问题。试验中大多数患者在治疗第三个周期时已无法维持最初的计划剂量,需要频繁调整剂量。而这可能进一步影响了药物的疗效和耐受性。
尽管GSK 对Blenrep (belantamab mafodotin-blmf)的获益/风险状况充满信心,并将继续与 FDA 密切合作,完成对复发或难治性多发性骨髓瘤患者使用Blenrep的审查,但在Blenrep撤出市场的两年多时间中,r/rMM的用药环境的剧变,两款BCMA CAR-T也成功从4线一跃至2线。另外,三款BCMA/CD3双抗在此期间相继获批,Blenrep的前景不容乐观。
Columvi:临床证据不足,美国人群比例低
7月18日,罗氏及其子公司基因泰克(Genentech)宣布,美国食品药品监督管理局(FDA)拒绝批准Columvi联合化疗方案GemOx(吉西他滨+奥沙利铂)用于治疗不适合自体干细胞移植的二线DLBCL患者。
罗氏Columvi(glofitamab)是一款靶向CD20/CD3的双抗,旨在通过同时激活T细胞与靶向B细胞来治疗复发/难治性弥漫大B细胞淋巴瘤(DLBCL)。

此次sBLA是基于III期STARGLO研究的结果。结果显示,中位随访11.3个月时,与利妥昔单抗联合GemOx方案(R-GemOx)组相比,格菲妥单抗联合GemOx方案组患者的总生存期(OS)实现了具有统计学意义和临床意义的延长(尚未达到
vs 9个月,HR=0.59,P=0.011);中位随访20.7个月时,格菲妥单抗联合GemOx方案组患者的OS获益保持(25.5个月 vs
12.9个月,HR=0.62)。
根据完整回应函,Columvi的3期Starglo试验数据未能为该药物在二线DLBCL美国患者群体中的应用提供足够的证据。这一决定与近期FDA咨询委员会会议的投票结果不谋而合,当时一个外部专家小组以8比1的压倒性票数认为Starglo试验的结果不适用于美国患者。
该研究在亚洲、澳大利亚、欧洲和北美的13个国家的62个中心纳入了274例患者,其中亚洲和澳大利亚人群占比59%,北美人群占比9%,欧洲人群占比32%。Starglo仅招募了9%来自美国的患者这一事实也引起了FDA的注意。罗氏公司给出了几种解释,包括西方国家更容易获得更有效的后续疗法,以及由于新冠疫情导致美国患者入组速度缓慢。但FDA及其咨询小组显然并不信服。
尽管Starglo的总体表现为积极,但FDA仍强调亚洲和非亚洲地区患者生存数据存在不平衡。根据FDA亚组分析,虽然在整个Starglo试验人群中,Columvi-GemOx 比Rituxan-GemOx 显著降低了41%的死亡风险,但非亚洲国家中,Columvi 方案的死亡风险高出6%。
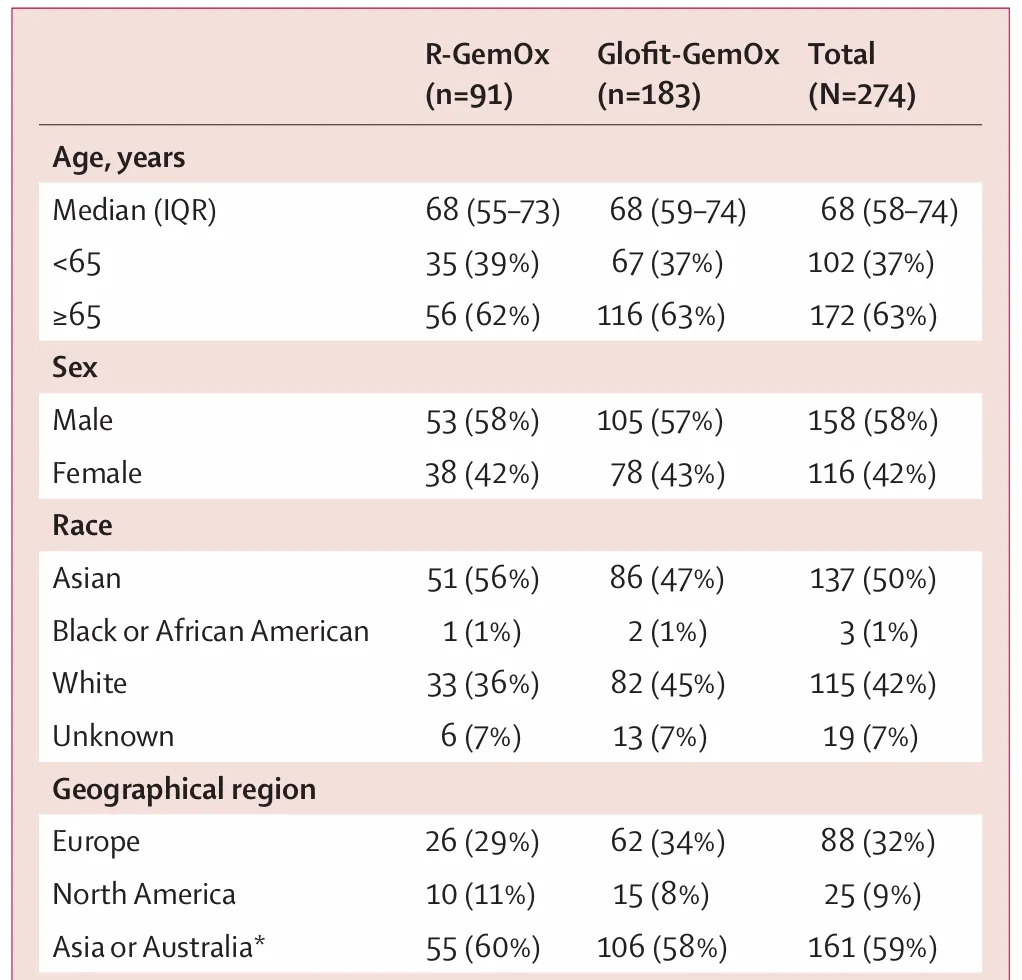
STARGLO也旨在作为一项上市后验证性研究,将 Columvi 在美国用于三线或以上 DLBCL 治疗的加速审批转化为全面审批。Columvi
仍处于三线或以上 DLBCL 患者的加速审批阶段。目前正在与 FDA 进行磋商,以确认 III 期 SKYGLO 研究作为新的上市后要求,该研究旨在研究
Columvi联合Polivy® ( polatuzumab vedotin-piiq)、Rituxan®
(利妥昔单抗)、环磷酰胺、阿霉素和泼尼松治疗既往未曾治疗的大B细胞淋巴瘤患者。
罗氏仍坚信Columvi在DLBCL第三线及以后治疗中的疗效及价值。同时,正在与FDA协商利用另一项三期研究SKYGLO作为后续确认性研究,SKYGLO评估Columvi联合Polivy(polatuzumab vedotin)和Rituxan为初治DLBCL患者的方案。
总结
两款药物被拒的共同痛点——“美国患者人群比例低,无法判断对美国人群的真正获益”。
众多业内权威专家纷纷发出呼吁,制药企业在推进临床研发工作的进程中,应当以更为精准、细致的方式,对全球患者参与临床试验的比例加以协调统筹。这不仅仅直接关联着新药能否顺利且快速地获得监管部门的批准,更深刻影响着广大患者用药安全性的保障程度。只有基于具有充分代表性的数据研发出的新药,才能最大程度地降低患者用药风险,让患者真正从新药中获益。
美国食品药品监督管理局(FDA)也多次着重强调,将持续不遗余力地推动美国本土患者在临床试验中的参与度提升。在此监管导向下,未来制药公司必须以更高的重视程度,精心规划并严格执行符合监管要求的临床试验设计。
 研发追踪
新药界 2026-05-21
455
研发追踪
新药界 2026-05-21
455
 研发追踪
药智数据 2026-05-21
570
研发追踪
药智数据 2026-05-21
570
 研发追踪
药番茄Pharmato 2026-05-18
592
研发追踪
药番茄Pharmato 2026-05-18
592
 热门资讯
热门资讯 微信公众号
微信公众号