

 产业资讯
产业资讯
 医药魔方
医药魔方  2025-09-16
2025-09-16
 1768
1768
9月10日,《纽约时报》发布报道称,特朗普政府考虑对来自中国的药品采取打击措施。该报道基于一项行政命令草案,涉及针对中美BD和数据监管的两大建议举措:
第一,对美国制药公司收购中国在研药物的交易进行更严格的审查,包括由美国外国投资委员会(CFIUS)进行评估;第二,要求FDA对中国临床试验数据进行更严格的审查,同时对提交中国试验数据的公司收取更高的监管费用。
关于加强跨境交易审查的建议,究竟能带来多大程度上的影响尚未可知。近两年越来越多中国创新药企选择走上交易驱动路线,地缘政治有一点风吹草动,都能在业界引发恐慌。不过,也有从业者分析,政治动作的风声或许会引发短暂的交易繁荣。
关于FDA对中国临床数据的认可度问题,诺华CEO Vas Narasimhan曾对此发表观点,“停止互认临床数据的风险确实存在,但如果全球临床试验区域化分裂,药物开发进程将严重受阻——仅美国不足以完成患者招募”,同时药企的研发成本也将增加。因此,上述建议若实施,对于美国药企来说同样意味着极大威胁。
暂且抛开未落地的“建议”不谈,对于中国药企来说,相对确定的是FDA审查将更加严格的趋势已现。
伴随全球供应链重构以及监管技术升级,FDA正通过一套 “组合拳” 重塑监管格局,不仅深刻影响全球制药业的竞争规则,更给积极出海的中国药企带来了前所未有的合规考验。面对国际环境日益紧张,中国药企现下能做的依然是保持严谨,在理解FDA监管变革逻辑的基础上增强竞争力。
FDA对海外临床态度:开放与警惕并存
在上述引发热议的报道中,《纽约时报》再次强调了特朗普政府寻求加快FDA审查流程的讨论。此前,FDA专员Marty Makary博士也提出了进一步加快药物审批速度的策略,比如国家优先审批券(CNPV)计划,但收到不少有关“偷工减料”和“审批政治化”的质疑声。
事实上,FDA确实曾因提升审批速度的牺牲了对数据量的要求。俄勒冈州立大学2023年的两项研究表示,FDA对数据较少的药物持开放态度。在2022年获批的37种药物中,有24种(65%)基于一项研究获得了批准,仅4种(约11%)在批准前报告了三项或更多研究。
不过患者招募问题仍然存在,大型药企发现仅美国患者很难满足临床试验需求,FDA不得不对海外临床数据更加开放。
CDER发布的《药物试验简报》显示,2024年,FDA批准了4种在临床试验中未纳入美国受试者的药物,涵盖治疗血液疾病、尿路感染、癌症以及一种罕见病药物,与2022和2023年的2种相比,数量有所提升。
从结果上来看,FDA越来越频繁地批准无美国受试者的药物,似乎正逐步降低对美国本土受试者数据的绝对依赖,更倾向于接纳多元化的海外数据,以缩短药物审批周期、满足患者对关键治疗药物的需求。
但对于中国药企来说,FDA对数据的种族多样性要求依然十分严格,同时对美国本土生物信息安全问题也越发警惕。
FDA曾表示,不会基于仅在中国境内开展的临床试验批准药物。2022年,FDA向礼来及信达生物发出完整回复函,要求为信迪利单抗补充美国患者群体数据。而去年获批的无美国参与者的药物,其临床试验也均在多个国家设有试验地点。
另一方面,明确表示提防中国和其他“关注国家”获取生物识别和基因组数据等敏感信息。例如,今年6月FDA宣布将停止“涉及将美国公民的活细胞送到中国和其他敌对国家进行基因工程,随后输回美国患者体内”的新研究。并表示正在审查相关试验,不会允许新试验继续进行。
但美国并没有让本土试验变得更容易。特朗普政府今年从NIH预算中削减了数十亿美元,预计将影响全美的临床资金,有从业者表示,美国药企依然在积极寻找全球其他地区临床试验的机会。
监管体系全方位升级,加强境外检查
地缘政治因素之外,FDA正迅速开展全方位监管体系升级——从 “事件驱动型合规审查” 转向 “持续化、全球化、数据驱动型监管体系”。
近半年来,FDA推出了三项重大变革,引领美国制药行业转型:
5月6日,受总统行政命令推动,FDA宣布扩大突击式境外检查的范围,旨在解决历史上国内和海外 “双重标准” 的问题;
6月26日,FDA发布最终指南《开展远程监管评估:常见问题解答》,正式将远程监管评估(RRA)纳入其永久性监管工具库;
7月10日,作为 “彻底透明化” 举措的一部分,FDA在其公开平台上推出了集中式数据库,包含200多份经过编辑处理的完整回复函(CRL);
此外,FDA还正式推出生成式人工智能工具Elsa,协助工作人员完成各类审查任务,例如识别高优先级的检查目标。
上述举措表明,FDA正在强化监管体系的严密和透明程度,并进一步强调了对数据完整性的关注。这些举措不仅提高了合规门槛,更改变了行业对FDA监管的传统认知——企业需从应对单次检查转向持续的合规准备。
为实现“随时随地”监管,FDA将RRA正式纳入监管工具库,并扩大境外突击检查范围,标志着监管向实时审查迈出重要一步。2025年6月26日发布的 RRA 最终指南,明确了在传统现场检查之外,RRA作为“独立常设监管机制”的地位,其适用范围也不仅限于药品生产质量管理规范(GMP),还包括药物临床试验质量管理规范(GCP)下的临床试验执行,以及所有其他良好规范(GxP)相关活动。
从技术层面而言,企业或生产场地可以“拒绝‘自愿性’RRA且不构成直接违规”,但指南明确指出,这种拒绝可能会“延误FDA做出监管决策的进程”(包括与待审申报材料相关的决策),或“促使FDA启动传统的现场检查”。这套整合体系使FDA能够通过远程审查电子记录,以低成本、快速的方式完成分类评估;若企业拒绝RRA或提供的数据不完整,FDA便据此启动突击检查。
“随时随地” 监管框架的另一重要组成是FDA增加对境外生产场地突击检查的频次,其核心目的是“要求境外制造商遵守与美国本土制造商相同的标准”,并更有效地“揭露那些可能通过提前通知检查而伪造记录、隐瞒违规行为的不良企业”。
同时也是为了回应国内药企质疑——早在新冠疫情前,FDA审查就开始受到指责,“对国内企业要求要严格得多,对海外企业却放宽了。”
审查趋严早有迹象,FDA发布的《2024财年药品质量状况报告》给出数据证明。自新冠疫情以来,FDA对国内外生产商开展的药品质量保证检查(包括监督性检查和有因检查)频次显著增加。总体上,2022财年为522次,2023财年为766次,2024财年为972次,逐年增加。
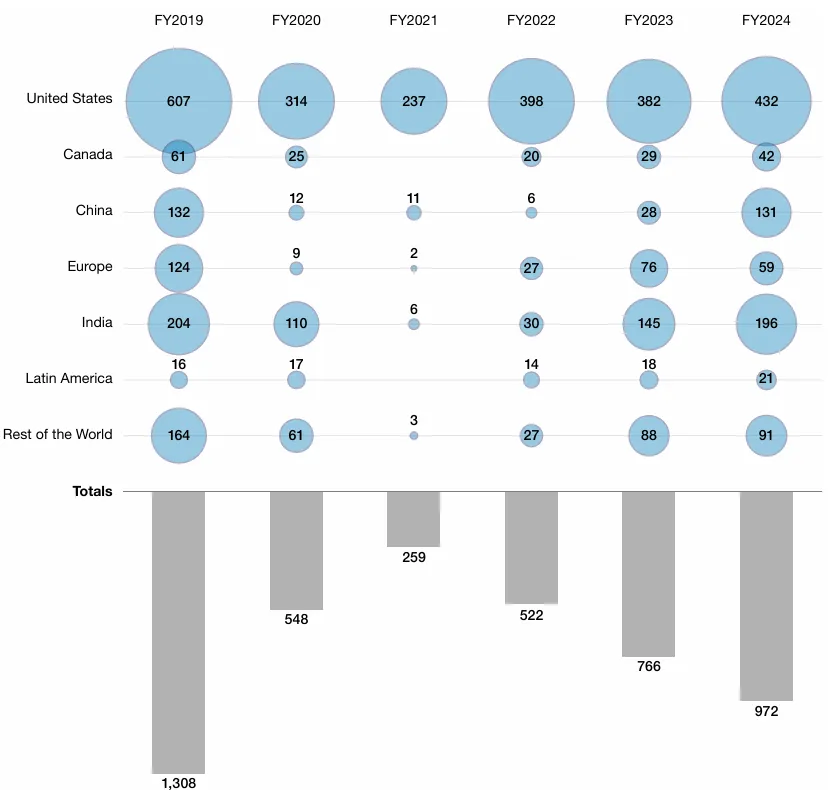
图1 2019财年至2024财年FDA按国家和地区分类的药品质量保证检查情况的项目
其中,2024财年FDA针对境外场地的药品质量保证检查达到历史最高水平,超过62%。其中印度和中国尤为显著:印度目录中34%的场地、中国目录中28%的场地均接受了检查。相比之下,2024财年美国场地目录中仅24%的场地接受了检查。
表1 截至2024财年,按国家/地区划分的、最新检查结果为NAI(无整改要求)或VAI(建议自愿整改)的场地比例
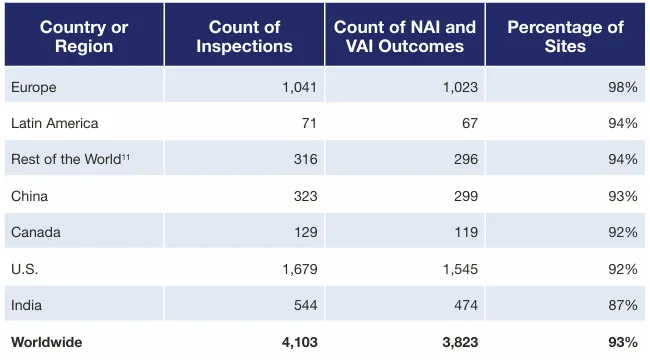
过往的行业实践已经证明,提升合规和质量要求是全球化竞争的长期通行证。CGMP检查结果可反映出各场地是否具备按FDA质量要求生产药品的能力,在FDA官网统计数据中,中国场地合格比例与全球平均齐平,甚至略高于美国。
综上,FDA审查趋严并非只受短期地缘政治波动影响,而是全球药品监管的长期趋势。FDA正在升级为更严格、更透明、更精准的监管体系,推动全球制药业提升质量标准,这也是中国药企当前能够把握的部分。
 产业资讯
摩熵医药 2026-05-07
135
产业资讯
摩熵医药 2026-05-07
135
 产业资讯
生物药大时代 2026-05-07
165
产业资讯
生物药大时代 2026-05-07
165
 产业资讯
药渡 2026-05-07
149
产业资讯
药渡 2026-05-07
149
 热门资讯
热门资讯 微信公众号
微信公众号