

 产业资讯
产业资讯
 Insight数据库
Insight数据库  2023-02-28
2023-02-28
 4872
4872
据 Insight 数据库统计,本周(2 月 19 日-2 月 25 日)全球共有 36 款创新药(含改良新)研发进度推进到了新阶段,其中 2 获批上市,3 款申报上市,6 款获批临床,7 款申报临床。
下文中,Insight 将分别摘取国内外部分重点项目做介绍。
国内创新药进展
国内部分,本周共有 38 款创新药(含改良新)研发进度推进到了新阶段,其中 5 款获批上市,1 款获批临床,11 款申报临床。
本周国内首次启动临床的 11 款创新药(含改良新)
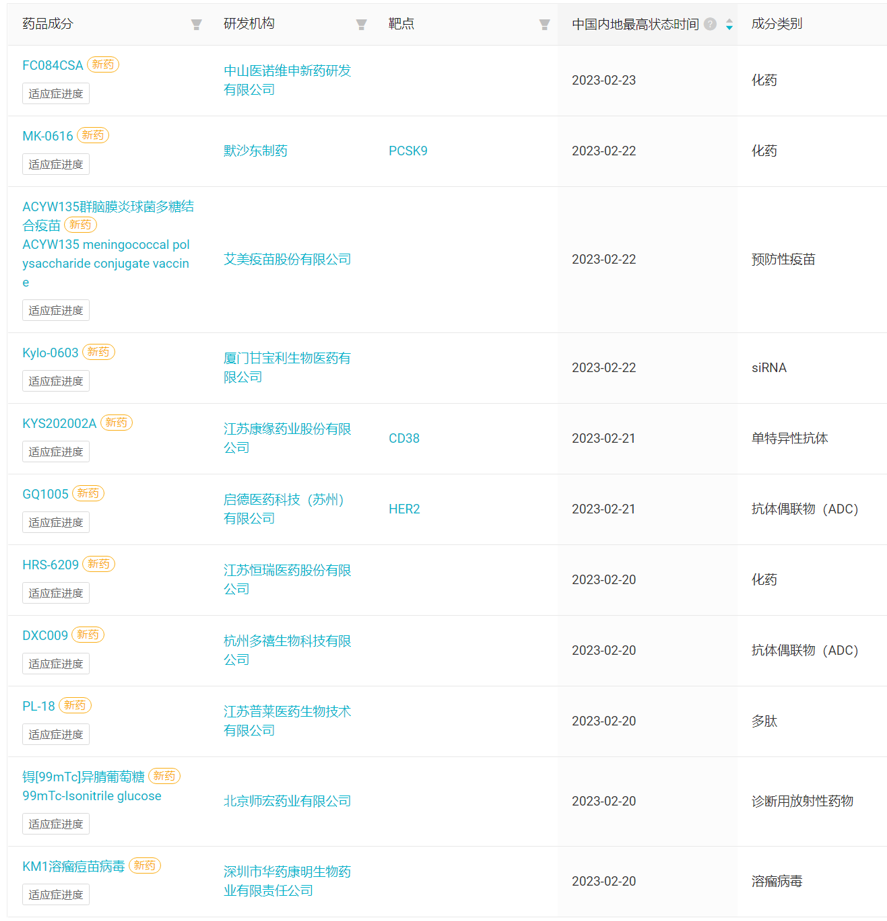
来自:Insight 数据库网页版(http://db.dxy.cn/v5/home/)
获批上市
1、第一三共/阿斯利康「德曲妥珠单抗」获批上市
2 月 24 日,NMPA 发布批件,第一三共/阿斯利康的重磅 HER2 ADC「德曲妥珠单抗」在中国首次获批上市(受理号:JXSS2200011),用于治疗既往接受过一种或一种以上抗 HER2 药物治疗的不可切除或转移性 HER2 阳性成人乳腺癌患者。
这项上市申请在去年 3 月 21 日首次在国内递交,如今获批上市,审评历时不到一年。值得一提的是,在此刻获得批准,意味着在 2023 年的新医保谈判中,德曲妥珠单抗大概率将参与。
不仅如此,该药用于 HER2 低表达(IHC 1+ 或 IHC 2+/ISH-)乳腺癌的新适应症也已经在去年 8 月申报上市(受理号:JXSS2200033),当前正在审评当中,预计将在下半年获批。
Enhertu 最早由第一三共开发,2015 年 9 月在美国和日本首次启动 I 期临床,迄今已经在 ClinicalTrails.gov 登记了 56 项临床试验,包括 11 项 III 期临床。试验地点遍布全球各地,为各主流监管机构的申报奠定了基础,带来了超过 40 个国家的获批,获批癌种包括乳腺癌、胃癌、非小细胞肺癌。
Enhertu 临床试验地图
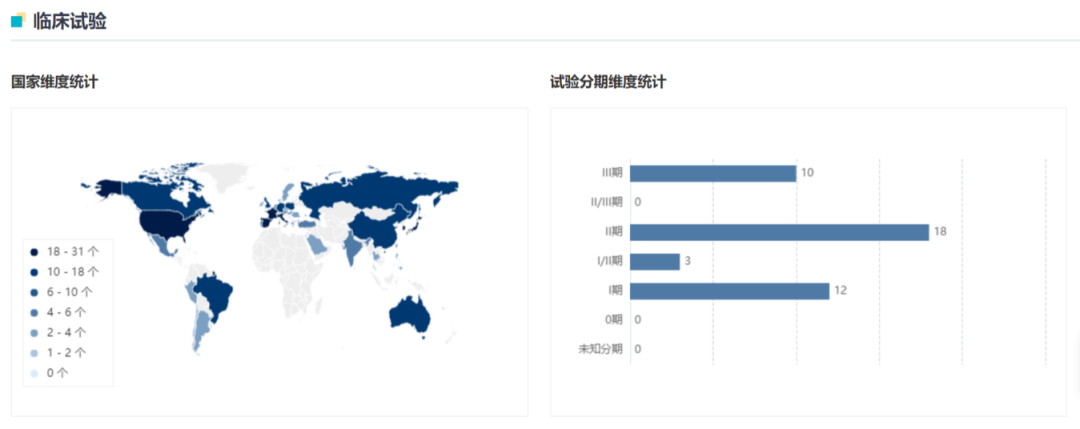
来自:Insight 数据库网页版(http://db.dxy.cn/v5/home/)2019 年 3 月 28 日,阿斯利康与第一三共签订全球合作开发和商业化协议,以首付款 13.5 亿美元 + 潜在里程碑 55.5 亿美元的金额大手笔获得 DS-8201;同年 12 月,基于 DESTINY-Breast01 研究(简称:DB01,下同),该药在美国获得 FDA 批准上市,用于三线及以后 HER2 阳性乳腺癌,商品名为 Enhertu。在日本,Enhertu 的首项批准发生在 2020 年 3 月,EMA 则在 2021 年 1 月。
第一三共和阿斯利康针对 Enhertu 的医药交易
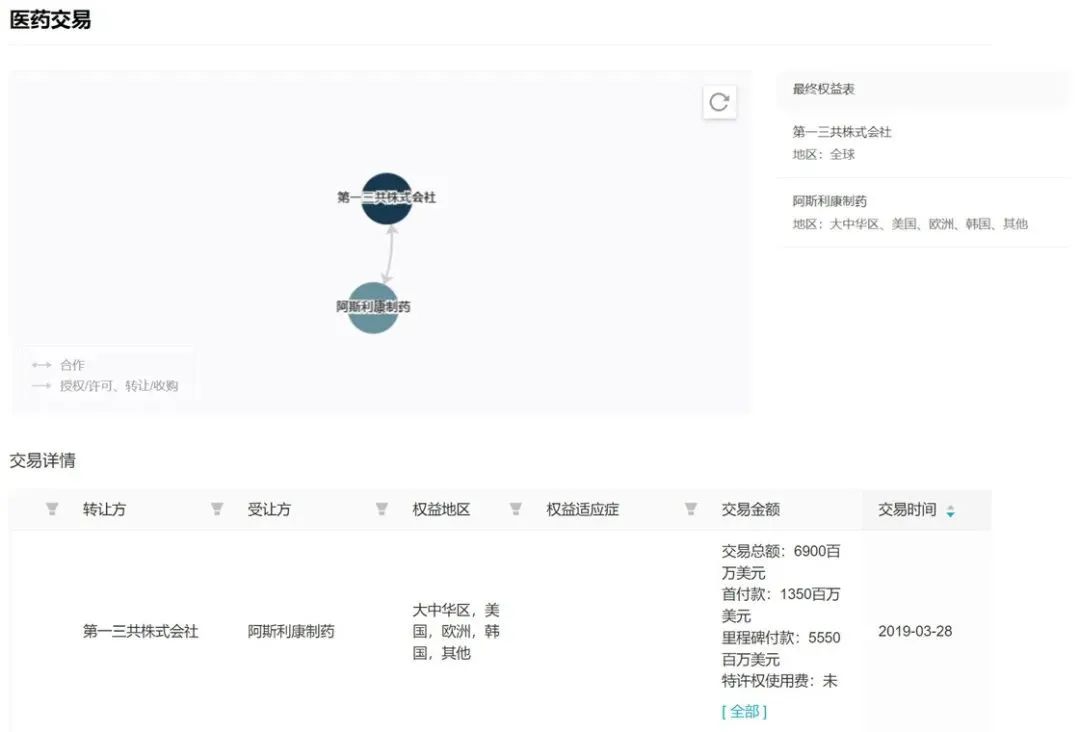
来自:Insight 数据库网页版(http://db.dxy.cn/v5/home/)获批上市以来,其销售额持续高速增长,尤其在各大新增适应症的持续渗透之下。
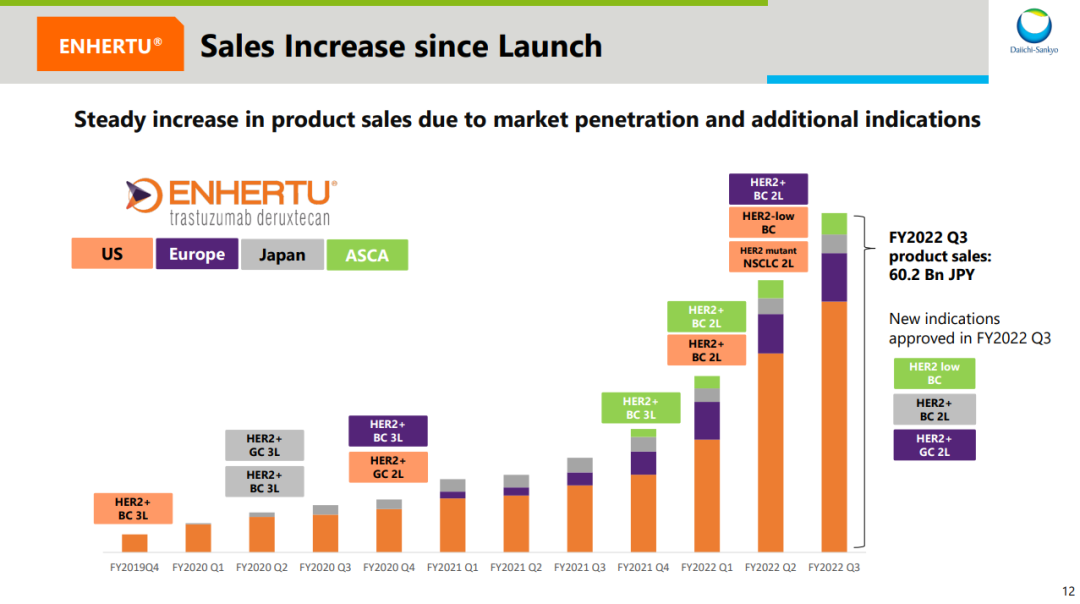
来自:第一三共 FY2022Q3 财报
Enhertu 自 2022 年度在 ASCO 上发布 DB04 临床结果之后,频频刷屏,成为药圈最亮眼的明星,直接影响了不仅 HER2 ADC 在内的一众企业在乳腺癌领域的布局。作为首个在 HER2 低表达乳腺癌中取得突破的疗法,Enhertu 定义了乳腺癌领域新的治疗标准,给既往被认为是 HER2 阴性的约 50% 患者提供了靶向治疗方案。
在乳腺癌患者中,约 20% 患者为 HER2 阳性(IHC 3+),50% 的患者为 HER2 低表达(IHC 2+/IHC 1+),15-30% 的患者 HER2 极低表达(0 < IHC < 1+),5 - 20% 的患者为 HER2 阴性。
乳腺癌中的 HER2 表达
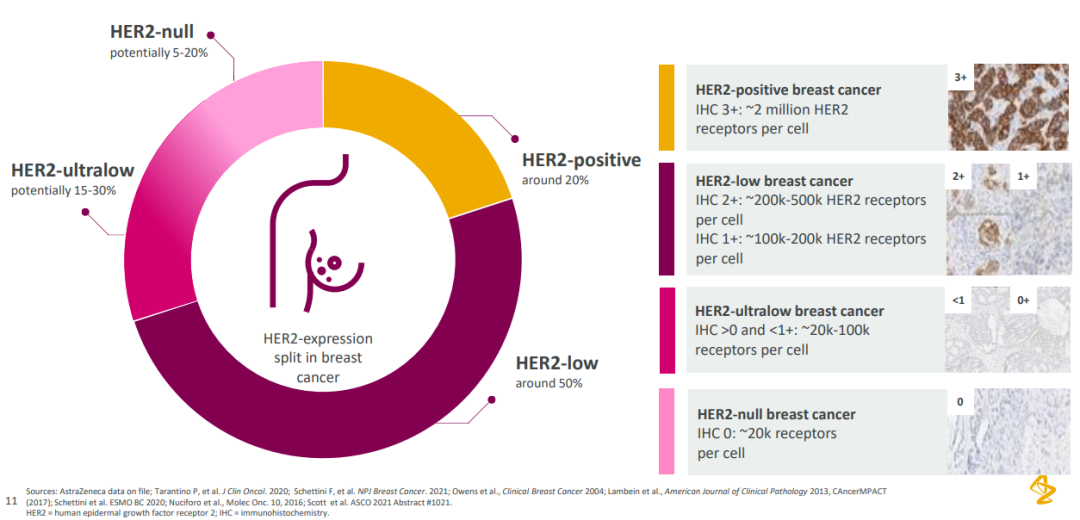
来自:阿斯利康 2022 ASCO 资料基于 HER2 低表达乳腺癌这一全新分型,第一三共的布局还不止于此。针对更前线治疗,DB06 和 DB08 研究都已经在推进中。前者是一项 3 期临床,针对 HR 阳性、HER2 低表达乳腺癌患者治疗;后者是一项联合疗法 1b 期临床,评估不限 HR 表达状态的 HER2 低表达乳腺癌患者,联用药物包括阿斯利康 PD-L1 度伐利尤单抗、SERD 氟维司群等等。
而其中,DB06 预计也将在今年上半年公布临床结果。据 Insight 数据库显示,DB06 研究的中国部分早在 2020 年 11 月就已经公示启动(登记号:CTR20202365)。
前线研究试验设计
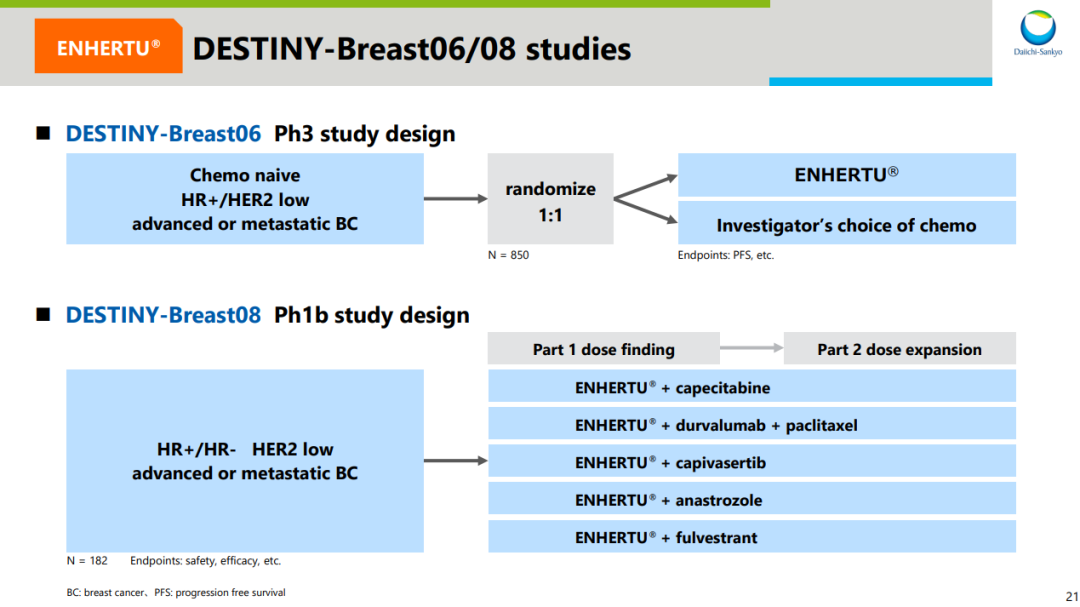
来自:企业官方资料
2、艾伯维:「乌帕替尼缓释片」第 4 个适应症国内获批上市!
2 月 20 日,NMPA 发布最新批件,艾伯维「乌帕替尼缓释片」的新适应症获批上市,用于治疗对一种或多种肿瘤坏死因子(TNF)抑制剂应答不佳或不耐受或禁忌的中度至重度活动性溃疡性结肠炎(UC)成人患者。这是该药获批的第 4 个适应症。
乌帕替尼缓释片(商品名:瑞福®),是一种选择性 JAK 抑制剂,目前正在数种免疫介导性疾病中对其进行研究。基于酶和细胞试验,乌帕替尼对 JAK1 显示出的抑制效力大于对 JAK2、JAK3 和 TYK2 的抑制效力。
在中国,乌帕替尼缓释片 15 mg 被批准用于治疗患有难治性、中度至重度特应性皮炎的 12 岁及以上的成人和儿童患者;用于治疗对一种或多种 TNF 抑制剂反应不充分或不耐受的中度至重度活动性类风湿关节炎成人患者;用于治疗对一种或多种 DMARD 反应不足或不耐受的成年患者的活动性银屑病关节炎;用于治疗对一种或多种肿瘤坏死因子(TNF)抑制剂应答不佳或不耐受或禁忌的中度至重度活动性溃疡性结肠炎(UC)成人患者。
在中国,治疗特应性皮炎、银屑病关节炎、溃疡性结肠炎、中轴型脊柱关节炎、克罗恩病和 Takayasu 动脉炎中的 3 期试验正在进行中。
本次批准是基于三项 3 期随机、双盲、安慰剂对照临床研究的有效性及安全性的数据支持, 也是乌帕替尼缓释片) 在胃肠病学领域中国获批的首个适应症。
两项诱导研究(U-ACHIEVE 和 U-ACCOMPLISH)采用的是乌帕替尼每日一次 45 mg,持续给药 8 周的方案,在随后的维持研究(U-ACHIEVE 维持研究)中使用 15 mg 或 30 mg,每日一次,持续至 52 周的给药方案。
在所有的临床试验中,相比于安慰剂,接受乌帕替尼治疗在第 8 周和 52 周时达到临床缓解的患者显著更多,这一主要研究终点是基于 mMS(改良的 Mayo 评分):排便频率亚评分(SFS)<1 且不大于基线,直肠出血亚评分(RBS)= 0, 内镜亚评分(ES)≤ 1 且无黏膜易脆。此外,研究还达到了所有次要终点,包括内镜改善和组织学-内镜黏膜改善(HEMI),以及维持期无激素临床缓解。与安慰剂相比,所有的主要终点和按次次要终点的 p 值均 <0.001。
3、吉利德:「安必速」在华获批
2 月 20 日,吉利德宣布,NMPA 已批准安必速®(AmBisome®,注射用两性霉素 B 脂质体,50 mg)上市许可申请,用于治疗侵袭性真菌病。安必速®已在全球范围内广泛使用 30 年,被誉为侵袭性真菌病治疗的「金标准」,其在国内的正式获批将为我国侵袭性真菌病患者提供一个新的经验性抗真菌治疗的一线选择。
安必速®是两性霉素 B 的小球型单层脂质体,是用于系统性真菌治疗的唯一真正脂质体。安必速®适用于发热、中性粒细胞减少的真菌感染患者的经验性抗真菌治疗;对传统两性霉素 B 的治疗无反应或者因为肾功能损害无法耐受的曲霉属、念珠菌属和/或隐球菌属感染患者;以及用于内脏利什曼病的治疗。
安必速®可全面覆盖念珠菌、曲霉、毛霉和隐球菌等多种真菌,其对念珠菌整体应答率更高达 90% ,且耐药罕见。得益于独特的靶向脂质体递送系统,安必速®能够显著提升药物安全性,有效降低不良反应发生率 ,其特殊代谢途径更能减少药物相互作用,即便对于血液科和重症监护病房内的肾功能不全患者,也无需调整剂量,在提升患者获益的同时,可有效降低护理负担,显著提升临床使用的便利性。
此前,借助博鳌特许药械进口政策优势,安必速®先行先试项目已在海南乐城启动,并成功救助了数位侵袭性真菌病患者,满足了临床急需。这款药物在中国的正式获批将有助于进一步满足我国临床的治疗需求,让广泛的高风险人群及早获益。
申报上市
罗欣药业:1 类新药新适应症报上市
2 月 20 日,罗欣药业宣布其消化系统疾病领域重点产品 1 类创新药替戈拉生片(商品名:泰欣赞®)新适应症十二指肠溃疡上市申请获得 CDE 受理。这将成为继反流性食管炎后,替戈拉生片递交的第 2 个适应症的上市申请。
替戈拉生片是中国首款自研钾离子竞争性酸阻滞剂(P-CAB),拥有全新的抑酸作用机制,通过竞争性地阻断 H+/K+-ATP 酶上的 K+结合位点而起到抑制胃酸分泌的作用。
和传统抑酸药物相比,替戈拉生片拥有 30 分钟快速起效、黏膜愈合率高、有效改善夜间酸突破、药物相互作用较少,且服药方便不受进食影响等显著优势。
目前,替戈拉生片治疗反流性食管炎的适应症已通过医保谈判,成功纳入新版国家医保目录。此外,替戈拉生片后续适应症也处于研发阶段,联合适当抗生素根除幽门螺杆菌(Hp)适应症正在开展 III 期临床试验;替戈拉生注射剂已启动研发,目前全球范围内尚无同类注射剂产品上市。
此次上市申请是基于替戈拉生片在国内开展的一项多中心、随机、双盲、双模拟、平行分组、活性对照的 III 期临床研究,由首都医科大学附属北京友谊医院张澍田教授牵头,旨在对比评价替戈拉生片与兰索拉唑在治疗中国十二指肠溃疡患者中的疗效和安全性。
试验共入组 400 例患者,经过 4 周或 6 周的治疗,证实替戈拉生片 50 mg 每日一次治疗中国十二指肠溃疡患者 6 周累计内镜溃疡愈合率非劣效于兰索拉唑 30 mg,显著缓解患者的胃肠道相关症状,且安全性和耐受性良好。
临床试验结果披露
1、针对乳腺癌,君实特瑞普利单抗联合化疗 III 期研究结果积极,即将报上市
2 月 20 日,君实生物宣布,特瑞普利单抗联合化疗治疗晚期三阴性乳腺癌的 III 期临床研究(TORCHLIGHT 研究)已完成方案预设的期中分析,独立数据监查委员会(IDMC)判定研究的主要终点达到方案预设的优效界值。君实将于近期与监管部门沟通递交该新适应症上市申请事宜。
TORCHLIGHT 研究(登记号:NCT04085276)是国内首个在晚期 TNBC 免疫治疗领域取得阳性结果的 III 期注册研究。这项随机、双盲、安慰剂对照、多中心的 III 期临床研究,旨在首诊 IV 期或复发转移性 TNBC 患者中比较特瑞普利单抗联合注射用紫杉醇(白蛋白结合型)与安慰剂联合注射用紫杉醇(白蛋白结合型)的疗效和安全性。
据本研究期中分析结果,与注射用紫杉醇(白蛋白结合型)相比,特瑞普利单抗联合注射用紫杉醇(白蛋白结合型)用于首诊 IV 期或复发转移性 TNBC 患者可显著延长 PD-L1 阳性人群的无进展生存期(PFS),同时,全人群和 PD-L1 阳性人群的次要终点——总生存期(OS)也显示出明显获益趋势。特瑞普利单抗安全性数据与已知风险相符,未发现新的安全性信号。关于详细的研究数据,君实生物将在近期的国际学术大会上公布。
NCT04085276 试验历史时光轴
据统计,全球乳腺癌的年新发病例数达 226 万,死亡病例数达 68 万,是全球发病率最高的癌症。在我国,乳腺癌年新发病例数达 42 万,死亡病例数达 12 万,分别占全球例数的 18.4% 和 17.1%。其中,三阴性乳腺癌(TNBC)约占所有乳腺癌的 15-20%,具有侵袭性强、复发率高和预后较差的特点。晚期 TNBC 对靶向治疗和内分泌治疗不敏感,缺乏特异性的治疗方法。
近年,以 PD-(L)1 抑制剂为代表的肿瘤免疫治疗药物在多个瘤种当中取得了一系列突破。据 Insight 数据库显示,全球已有 19 款 PD-(L)1 抑制剂获批上市,这其中多数均已在布局乳腺癌领域,详见下图:
全球已获批上市 PD-(L)1 乳腺癌适应症研究进展(II 期临床及以上)
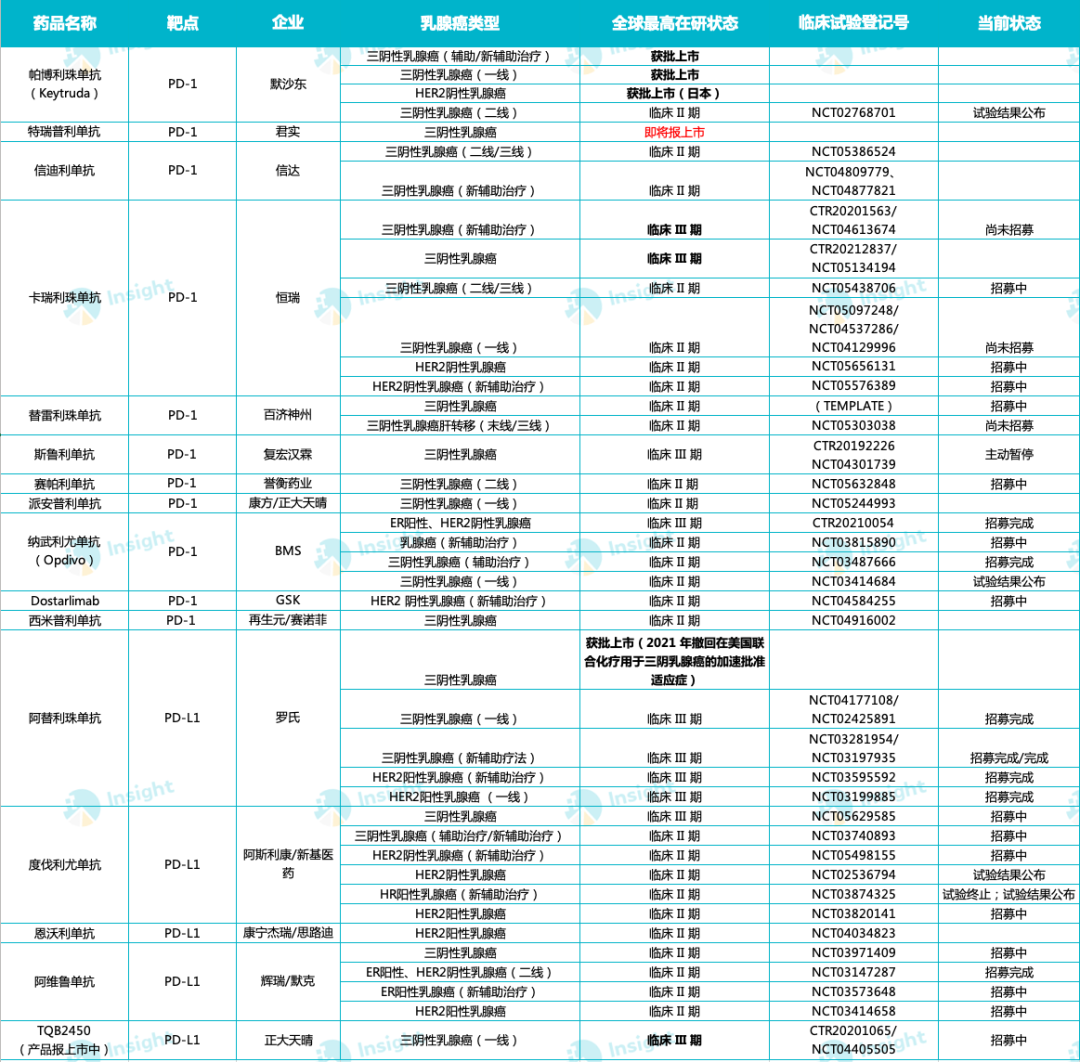
来自:Insight 整理(人工整理,如有纰漏请指正)然而,当前仅有默沙东 K 药以及罗氏 Tecentriq 获批乳腺癌适应症。其中,K 药获批的三阴乳腺癌适应症疗法线数及类型已覆盖一线、新辅助以及术后辅助疗法。对于后者而言,罗氏 Tecentriq 在该适应症的道路就并非如此顺利了。
2021 年 8 月,罗氏宣布自愿撤回 T 药联合化疗用于 PD-L1 阳性不可切除局部晚期或转移性三阴乳腺癌(mTNBC)的加速批准适应症。该决定仅影响美国的 mTNBC 适应症,不影响 Tecentriq 在美国和美国以外包括 mTNBC 在内的其他批准适应症。
对于国产药而言,当前并未有 PD-(L)1 抑制剂获批乳腺癌适应症,特瑞普利单抗是国内首个在晚期 TNBC 免疫治疗领域取得阳性结果的 III 期注册研究。此外,恒瑞卡瑞利珠单抗以及正大天晴的 TQB2450 也正在开展 III 期临床。
境外创新药进展
境外部分,本周共有 11 款创新药(含改良新)研发进度推进到了新阶段,其中 1 款获批上市,3 款申报上市,7 款获批临床。
获批上市
1、一线肝癌和 NSCLC!阿斯利康 PD-L1+ CTLA4 联合疗法获欧盟批准
2 月 22 日,阿斯利康发布公告,PD-L1 单抗度伐利尤单抗(Imfinzi)、CTLA4 单抗替西木单抗(Imjudo)联合疗法 2 项适应症获欧盟批准:晚期或不可切除肝癌(HCC)成年患者的一线治疗,以及与化疗联用治疗 IV 期(转移性)非小细胞肺癌成人患者的治疗。
HCC 适应症的获批是基于 HIMALAYA III 期试验的积极结果。这是一项随机、开放标签、全球多中心 III 期临床,评估了 Imfinzi 单药、Imfinzi 联用 CTLA-4 单抗 tremelimumab 与索拉非尼相比一线治疗肝细胞癌的疗效。
Imfinzi 和 Imjudo 联合疗法使用被称为 STRIDE 方案的给药方式,单次给药 Tremelimumab(300 mg)+ 高剂量 Imfinzi(1500 mg),随后每 4 周一次给药 Imfinzi。
在 2022 ASCO GI 中,阿斯利康公布了 HIMALAYA 研究的最终数据。相较于索拉非尼组,接受 STRIDE 方案治疗的患者死亡风险降低了 22%(HR = 0.78;96.02%[CI]:0.65-0.93,p = 0.0035)。联合疗法组中位 OS 为 16.4 个月,而索拉非尼组为 13.8 个月。据估计,31% 接受联合疗法的患者在 3 年后仍然存活,而索拉非尼组这一数值为 20%。
HIMALAYA 研究疗效数据
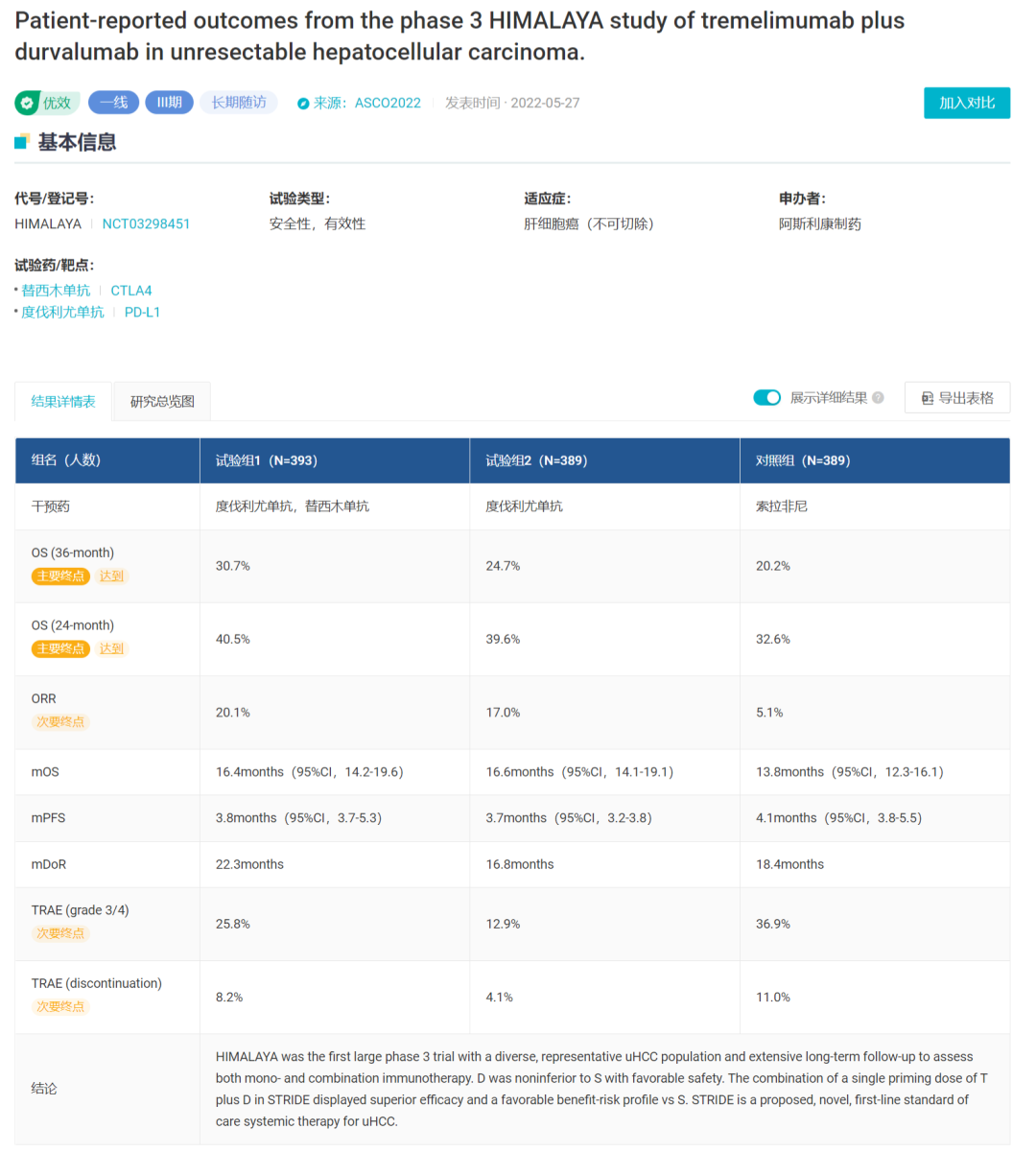
来自:Insight 数据库网页版而获批 NSCLC 适应症是基于 III 期 POSEIDON 试验的结果。
与单独化疗相比,Imfinzi 联合四个周期的化疗中加入有限疗程的 Imjudo,可显著降低 23% 的死亡风险 (HR = 0.77;95% CI:0.65-0.92;p = 0.00304),中位 OS 为 14.0 个月,化疗为 11.7 个月。据估计,33% 接受该联合疗法的患者在两年内仍能存活,而化疗患者的存活率为 22%。
与单独化疗相比,这种联合治疗还降低了 28% 的疾病进展或死亡风险 (HR = 0.72;95% CI:0.60-0.86:p = 0.00031),中位无进展生存期(PFS)分别为 6.2 个月和 4.8 个月。
POSEIDON 试验结果

来自:Insight 数据库网页版在 2022 ESMO 大会上发布的 POSEIDON III 期临床试验的更新结果显示:
与单独化疗相比,在 Imfinzi 联合铂类化疗的基础上再联合 5 个周期有限疗程的 tremelimumab(Imjudo)可将总生存期提高 25%(HR = 0.75;95% CI: 0.63-0.88)。联合治疗组更新的中位 OS 为 14 个月,而单独化疗组为 11.7 个月。接受联合治疗的患者中据估计有 25% 在三年时仍然存活,而这一比例在接受单独化疗的患者中仅为 13.6%。
POSEIDON 试验更新结果

来自:Insight 数据库网页版
申报上市
辉瑞疫苗获 FDA 优先审评资格,预防婴幼儿常见感染
2 月 22 日,辉瑞(Pfizer)宣布,FDA 接受其呼吸道合胞病毒(RSV)候选疫苗的生物制品许可申请(BLA),并授予优先审评资格。本次上市申请是为了通过怀孕妇女产生主动免疫,进而预防婴幼儿自出生起至六个月出现 RSV 引起的下呼吸道疾病。FDA 预计在 2023 年 8 月完成审查。若获批,此疫苗将会成为使用于怀孕妇女,以避免婴孩出生后感染 RSV 相关疾病的首款疫苗。
RSVpreF(PF-06928316)为在研的 RSV 疫苗,是根据美国国立卫生研究院(NIH)所建立的 RSV 融合前(prefusion)F 蛋白晶体结构所制造。此融合前蛋白为 RSV 病毒用以进入人体细胞的 F 蛋白的主要形态。NIH 的研究显示靶向此融合前蛋白形态可以有效阻断病毒感染。此双价疫苗含有等量、分别来自 A 与 B 病毒亚型的重组 RSV 融合前 F 蛋白。此疫苗于 2022 年 3 月时获得美国 FDA 的突破性疗法认定(BTD),用于怀孕妇女接种以避免 0-6 个月婴孩感染由 RSV 引起的相关下呼吸道疾病。
这次的申请主要是基于 MATISSE 临床 3 期试验的积极顶线数据。
预定中期分析显示,MATISSE 试验达成两个主要终点的其中一项。即疫苗避免婴孩出生后 90 天内感染严重性 MA-LRTI 的保护力达 81.8%(CI:40.6-96.3%),避免婴孩在出生后 6 个月的追踪期间感染严重性 MA-LRTI 的保护力达 69.4%(CI:44.3-84.1%)。虽然未达成统计显著水平,但仍可在第二项主要终点的分析上,见到疫苗的效力。即疫苗避免婴孩出生后 90 天内,预防 MA-LRTI 感染的保护力达 57.1%(CI:14.7-79.8%),避免婴孩在出生后 6 个月的追踪期间内感染 MA-LRTI 的保护力达 51.3%(CI:29.4-66.8%)。
在试验期间内预定,由外部数据监测委员会(DMC)所进行的定期安全性检验,显示此在研疫苗具良好的耐受性,对接种的妇女与其婴孩没有安全上的顾虑。
多说一点
CLDN18.2 靶点第 7 笔 License out 交易!康诺亚 ADC 牵手阿斯利康
2 月 23 日,康诺亚宣布已与阿斯利康达成交易,将 CLDN18.2 ADC 项目 CMG901 的研发、注册、生产和商业化全球独家权益授权给 AZ。交易总额包括 6300 万美元首付款、11.25 亿美元潜在里程碑款以及基于净销售额的分级特许权使用费。
 产业资讯
珍立拍 2026-05-19
430
产业资讯
珍立拍 2026-05-19
430
 产业资讯
深究科学 2026-05-19
430
产业资讯
深究科学 2026-05-19
430
 产业资讯
中国医药报 2026-05-19
557
产业资讯
中国医药报 2026-05-19
557
 热门资讯
热门资讯 微信公众号
微信公众号