

 产业资讯
产业资讯
 行舟Drug
行舟Drug  2023-06-16
2023-06-16
 8828
8828
来源
《中国新药杂志》 2023年第32卷第10期
作者
贾国舒,王敏慧,梁毅
中国药科大学国际医药商学院
摘要
自2017年我国加入国际人用药品注册技术协调会(ICH)之后,逐渐加强了境外临床试验数据衔接方面的研究,桥接研究的规范化和国际多中心临床试验将成为大势所趋。在境外已上市境内未上市药品注册过程中,境外临床试验数据的外推与衔接尤为重要。通常国家药品监督管理局药品审评中心(CDE)需要考量临床试验数据包的完整性,判断该药物是否具有种族敏感性、是否需要进行桥接研究。目前针对种族因素对境外临床试验可接受性的影响研究,国际上普遍遵循ICH E5(R1)技术指导原则,另外美国、日本、欧盟等已出台各自有关的法规或指南文件。本文介绍了我国当前对于境外临床试验数据审查的依据和要点,根据国际经验对桥接研究的要点进行了汇总分析,最终对我国境外临床试验数据衔接提出合理性建议。
关键词
境外临床试验;桥接研究;国际多中心试验;国际人用药品注册技术协调会
正文
随着临床研究的国际化,国外临床试验数据用于国内药品注册的情况逐渐增加,在我国通常表述为“境外临床试验数据的接受”。“境外”指关境,即中国港澳台地区临床试验也纳入“境外临床试验”的范畴。而对于美国、日本等国家,“境外”通常指国境,因而其法规与指南通常表述为“国外临床数据”的衔接。
为了评估种族因素在特定用法用量下对药品安全性和有效性的影响,基于种族因素合理设计用药方案,并最大限度地避免临床试验的国际重复实施,加快进口创新药与改良型新药在我国的注册进程,科学地引入境外临床试验数据显得尤为重要。而当境外的临床试验数据无法满足审查要求时,通常需要增加桥接研究以引入境外临床试验数据的部分或全部。
创新药在我国申报上市通常需要经过新药临床研究申请(investigational new drug,IND)。而跨境临床试验包括“IND境内临床试验”、“IND境外临床试验”、“非IND境外临床试验”中的1种或几种。临床试验申请人可以选择是否通过IND进行境外临床试验——若进行IND,则须在IND要求下开展研究;若不进行IND,申办者须确保临床研究满足《中华人民共和国药品管理法》和《药品注册管理办法》的要求,满足IND或新药上市申请(new drug application,NDA)的要求。当申办人仅使用境外临床试验数据进行新药上市申请时,鼓励其与国家药品监督管理局(NMPA)药品审评中心(CDE)人员开展预申请会议进行协商。
1 境外临床试验数据的审查
1.1 审查依据:ICH E5(R1)指导桥接研究
1998年3月,ICH出台了关于接受国外临床资料的种族影响因素要求的ICH E5(R1)指导原则。ICH E5指导原则讨论了如何将境外临床试验数据外推到境内的新地区,使既有的药物临床试验数据用于新地区的注册审批。
如果境外临床试验数据符合境内注册审批要求,且包含来自新地区的足够样本量的受试者,就可对种族差异进行初步分析。当境内外两试验地区间可能存在内在或外在的种族差异,或来自新地区的受试者数量不满足要求时,则须借助桥接研究使现有临床数据外推至新地区人群[1]。
我国境外临床试验数据审查流程见图1。
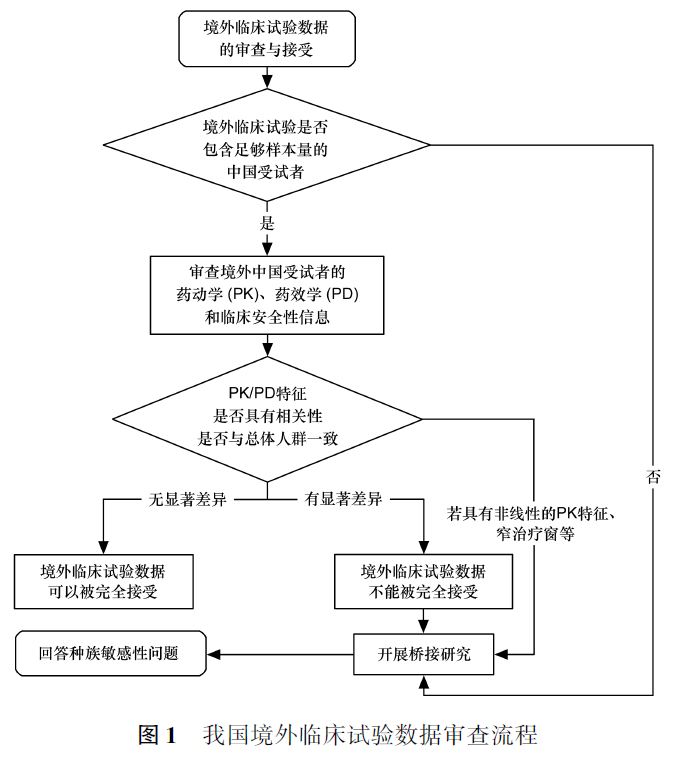
例如,某药物对种族因素较为敏感,其用法用量可能因地区而异,则需由新地区的监管部门判断原有效性研究结果是否可以外推至新地区——首先,判断原试验地区的数据是否满足新地区的注册审批要求,如不满足则需要进行桥接研究;其次,考察在新地区进行的桥接研究是否支持原试验地区的结论,如结论一致则无需进一步研究。但新地区的桥接研究未必遵循与原地区完全相同的剂量和疗效,可以在提供了相应安全性资料的情况下调整剂量和疗效指标。
但对于桥接研究所应包含的内容——药动学/药效学(PK/PD)研究、剂量⁃反应研究或是完整的临床试验,ICH E5并未作出规定。
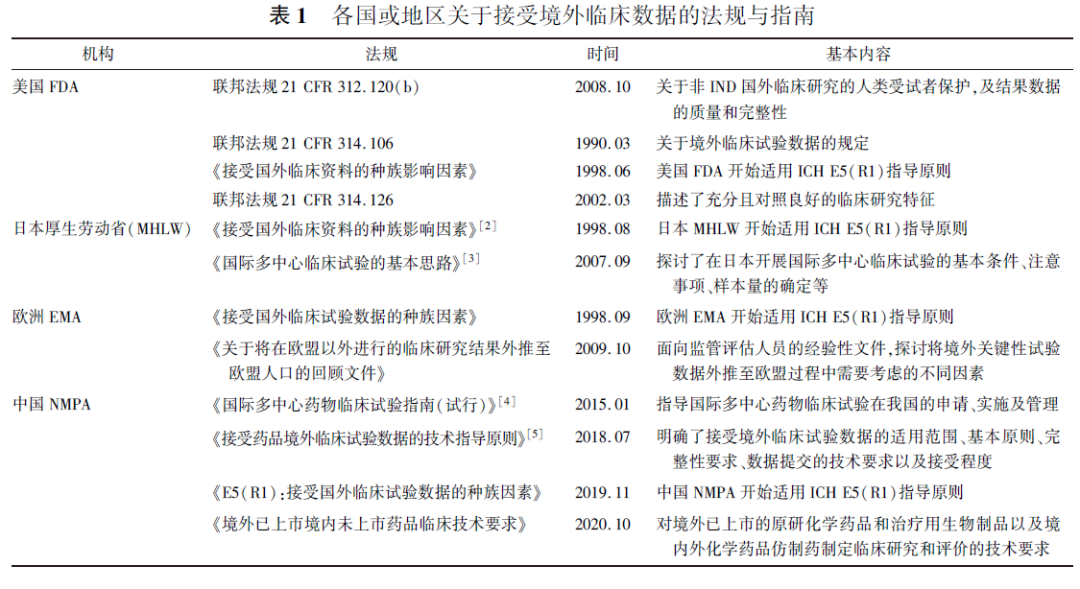
各国或地区关于接受境外临床试验数据的法规指南见表1。
1.2 审查要点:临床试验数据包的考量因素
若某药品已在某ICH成员国获批,当在中国提交注册申请,且预估临床试验数据与桥接研究数据可被CDE接受时,则应当向相关部门提供相应的证据予以支持。申请人需要向监管机构证明:现有数据能够充分满足新地区的监管要求,且适用于新地区人群。如果原试验地区的对照组、主要临床终点、纳入标准、诊断标准等关键临床试验设计特征不适用于新地区,则应向新地区的监管部门说明桥接研究的设计思路。
简言之,首先,需要说明现有的临床试验数据足以证明药物的安全性和有效性;其次,需要证明在新地区进行的桥接研究是必要的,并出具明确的方案。
1.2.1 科学评价临床试验数据
某药品已在境外上市或已在境外开展临床试验,应当具有真实、准确、完整、可追溯的临床试验数据,主要包括用于注册的临床试验数据和上市后的临床试验数据[6],其在境内上市时,须考虑数据包中的境外临床试验数据是否适用于境内,即监管机构需要对临床数据包进行审查。
该临床数据包应当包含:①PK数据,主要来源于药物的吸收、分布、代谢、排泄相关的试验。②PD数据。③剂量⁃反应数据。④桥接研究数据。
其中,①②③可将境外数据沿用至境内,④为境外研究外推至境内所收集的数据[7]。
以上明确表明剂量⁃反应、有效性和安全性的临床数据,须遵守新地区的标准和国际通行药品临床试验管理规范(Good Clinical Practice,GCP)的规定,并具有适当的对照和良好的管理。
对于境外已上市而境内未上市的药品,其临床试验要求见表2。

1.2.2 协商开展桥接研究
在境外临床试验数据包的基础上,由于地区间可能存在种族差异,境外的临床数据未必适用于境内人群,有时需要在新地区补充验证安全性、有效性、用法用量及药动学特性的桥接试验,才能构成完整的临床试验数据包。并且,当原地区和新地区的疾病发生率差别较大,而药物使疾病发生率降低的幅度相同时,为了保证该药物的风险⁃获益平衡,有必要补充临床试验以对其进行评估。
由于ICH E5(R1)指导原则没有针对特定地区作出规定,因此申请人应当在与CDE协商后确定桥接研究的计划。原则上,CDE应在认定境外临床试验数据包的完整性之后,再与申请人协商决定桥接试验的种类和规模。但也存在数据包尚未完成便与监管机构协商桥接研究设计的情况[8]。
同时,也存在可以豁免桥接研究、仅收集PK数据的情况——药物的性质不易受种族因素影响,且与种族因素的外在因素相似的情况;原试验地区和新试验地区种族组成相似,且两地区内许多药物不存在种族差异的情况。
1.2.3 种族敏感性的考量
药物种族因素是否敏感是开展桥接试验与否的重要考量因素。
“对种族因素敏感的药物”的消除代谢通常有遗传多态性酶参与,剂量⁃反应曲线斜率较陡峭[9]。
而“对种族因素不敏感的药物”具有以下特征中的1种或几种[4]:①具有线性药动学特征。②治疗剂量范围较宽,剂量⁃反应曲线斜率较平坦。③治疗范围广。④少量代谢或通过多种途径代谢。⑤代谢没有遗传多态性的酶参与。⑥生物利用率高,药物吸收不受饮食影响。⑦血浆蛋白结合率低。⑧药物相互作用的可能性较小。⑨作用是局部的。⑩不合理用药的可能性较低。
1.2.4 适应证的衔接
如果原试验地区针对某药物批准了多个适应证,即使主要适应证的桥接数据可外推至新地区,其他适应证也需考虑桥接研究的可能性,与新地区监督管理部门进行充分协商,不能一概而论。
若该适应证只是主要适应证的扩展,则可以考虑免除桥接研究;若该适应证对于新地区来说是新用途,则需要进行桥接研究。
1.2.5 特殊人群临床试验数据的衔接
如果某桥接研究已解决了该药物在普通人群中的安全性、有效性、剂量等问题,并在特殊人群(如肾脏/肝脏损伤患者、老年人、儿童、孕妇和哺乳妇女)中投入使用,如果特殊人群的研究设计同时满足CDE要求,则无需再探讨特殊人群临床数据的外推可行性。然而,对于特殊人群(如儿科抑郁症)的新适应证则需要进行单独的桥接研究。
2 桥接研究要点
2.1 桥接方案的设计
根据日本学者内藤周幸[10]的研究,进行桥接研究外推临床试验数据可分为以下5种情况:①将境外进行的Ⅲ期临床试验外推到境内的情况(此种情况最为常见)。②同样是将境外的Ⅲ期临床试验外推到境内,另外兼做剂量确认试验的小规模Ⅲ期临床试验的情况。③境外处于Ⅱ期临床试验后期,将结果外推到境内时需要在境内进行PK试验,并将药理学指标与临床效果指标进行关联的情况(如某些抗恶性肿瘤药)。④境外处于Ⅲ期的长期安全性试验,需要比较境内外进行的Ⅱ~Ⅲ期临床试验期间发生不良事件的种类和发生率,以及Ⅲ期和长期安全性试验不良事件的情况——可在境内实施Ⅱ~Ⅲ期临床试验作为桥接试验,或将针对有效性进行的Ⅱ期/Ⅲ期临床试验作为桥接试验。⑤将境外进行的针对特殊患者人群的临床试验外推至境内的情况,在境内进行的Ⅱ期/Ⅲ期临床试验将作为桥接试验,但必须采用可进行比较的方案设计。
由于桥接研究的目的是在境外临床试验的基础上,尽可能不进行大量新研究,除了允许将临床试验数据外推到新地区,通常还会比较两地区的剂量⁃反应关系。因此,在实际研究中,桥接研究的时机、种类以及原临床试验数据包如何外推,都要有明确的试验方法和设计方案。
首先,须明确境外临床试验数据包中需要进行“搭桥”或外推的研究,选择适当的研究作为“桥梁”的基础,在新地区的研究应当采取与其相同或相似的方案设计——受试者、研究持续时间、终点指标以及临床效果评价指标等,原基础研究和新的桥接研究应当可以进行类比[10]。
另外,多区域研究应招募足够数量的受试者,以保证每个试验地区所得到的临床数据科学合理。允许地区研究设计方面存在一些微小差异,如受试者年龄纳入标准、受试者伴随用药等。
2.2 桥接研究的种类
一般,桥接研究可包括以下几种:①与药物安全性、有效性有关的临床试验。②与药物用法用量相关的临床试验。③PK试验。④PD试验。其中,①一般指Ⅲ期临床试验,②一般指Ⅱ期临床试验。
2.2.1 PK/PD研究的职能
桥接研究首先应评估两地区人群的PK曲线相似性,根据PK的种族差异决定是否需要调整用药剂量。因此,通常PK研究是必需的——可独立于桥接试验进行研究,也可在桥接研究中测量PK数据。
据中出進等[11]的研究,PK/PD研究可分为独立型和附加型2种,皆可应用于桥接研究。前者是将PK/PD研究作为独立的临床药理学试验,旨在获得有关PK/PD的详细信息,虽是小规模试验,但在药物研发初期可发挥重要作用。后者类似于Ⅱ期临床试验,是将PK/PD研究作为药物安全性/有效性试验中的一个附加环节,相比独立型研究可以节约费用与时间,也可纳入更多的受试人群。
2.2.2 剂量⁃反应试验
桥接研究主要需要获得PK数据、PD数据和剂量⁃反应数据,而相似性和外推性是桥接试验主要的验证对象,因此,剂量⁃反应试验是桥接试验的主体。剂量的设定方面,首先应优先选择和原试验地区相同的剂量,其次根据PK和PD研究结果适当调整,有时为确保安全性可能会取消某些高剂量组别的设置[8]。
对于药物在原地区和新地区之间临床意义上相似性的判断,通常可以采用剂量⁃反应曲线进行比较,另外还可根据不同剂量组别之间、安慰剂组别之间以及置信区间是否具有统计学意义上的显著性差异进行评估[12]。
2.2.3 桥接研究数据可接受性的判断
桥接研究数据能否外推至新地区,首先应判断该药物在新地区的剂量⁃反应、安全性及有效性与原试验地区是否具有相似性,若无显著性差异,则可作为境外临床试验的桥接研究;若原剂量不适用于新地区人群,而调整药物剂量后在新地区人群中所体现的安全性和有效性特征与原试验地区无显著性差异,则可通过PK/PD研究进行剂量调整,将境外临床试验数据外推至新地区。
另外,若桥接试验的规模与受试者数量不足以将境外不良事件外推至新地区人群,则应当补充安全性数据;若桥接试验不能验证药物的安全性和有效性,则需要补充临床研究数据[6]。
2.3 种族因素的判别
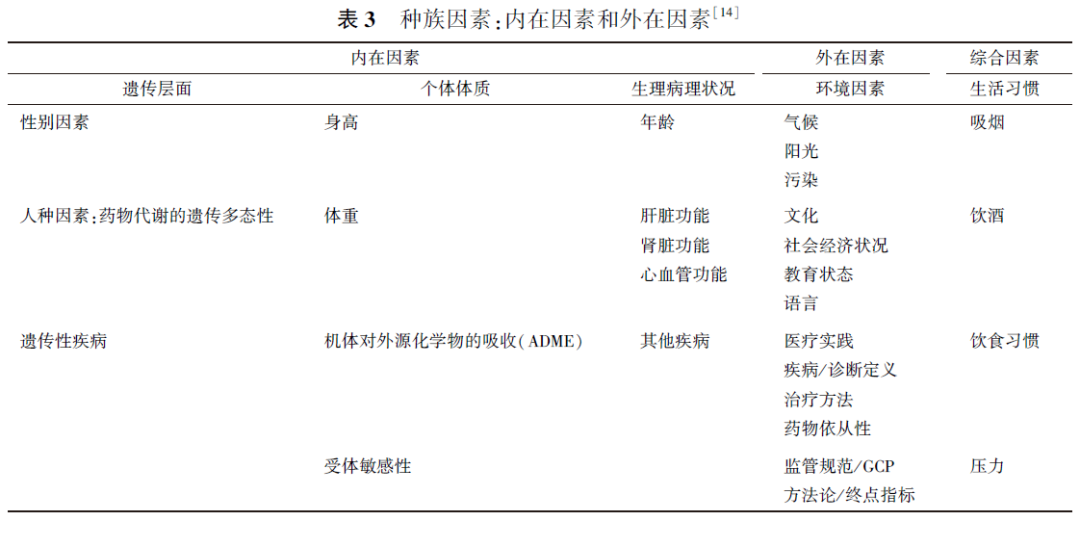
根据ICH E5原则[1],种族因素可分为内在因素和外在因素,内在因素包括遗传多态性、年龄、性别、身高、体重、瘦体重、机体成分、器官功能障碍等,其有助于定义一个亚群体,并可能影响地区间外推临床数据的能力;外在因素包括医疗实践、饮食、吸烟、饮酒、暴露在污染和阳光下的程度、社会经济状况、用药依从性,以及临床试验的设计和实施等,主要取决于受试者所处的文化与社会环境,临床研究对地区的依赖性越强,外在因素影响越大。种族的内在和外在因素见表3。
当原试验地区无新地区的足够受试者时,申请人应当通过桥接研究识别出地区间不同的内在因素,并表明这些因素不会对药物的有效性产生显著影响,即表明药物对种族因素的任何差异都不敏感。目前,在我国5.1类化学药审评审批时,通常会考虑药物在日本人和高加索人种中所体现出的PK和PD参数,以判断药物在我国是否具有种族敏感性[13]。
申请人还应尽可能利用数据证明药物在两地区间能够产生相似的临床效果。针对预期人群可能出现的外部因素,申请人应当证明其对药物的有效性不会产生显著影响,并说明原因。
3 关于境外临床试验数据衔接的展望
3.1 规范桥接研究的设计
目前我国5类新药仍是注册热点,而境外上市药品在申请境内上市时仍存在临床试验类型杂乱、没有统一桥接标准等问题,虽然中国接受了ICH E5原则,但相关研究较少,对于桥接研究开展的时机、研究种类、疗效终点指标、“相似性”指标及统计方法等尚未出台相关指南予以规范。
虽然临床试验数据只要能够充分论证药物在境内的安全性和有效性即可,但仍需要有统一的标准来界定是否需要进行桥接研究、桥接研究是否包括临床试验以及应包括的试验项目,这是目前我国在药品审评过程中没有明确规定的。
3.2 鼓励国际多中心临床研究
根据Kogure等[15]的研究,通过早期启动桥接研究(即在境外完成关键研究前启动桥接研究)或进行国际多中心临床研究都可缩短药物审评滞后时间。ICH E5原则的主题是判断境外临床试验数据的可接受性及如何进行衔接,而桥接方案的设计并非其研究重点。2020年10月,CDE出台《境外已上市境内未上市药品临床技术要求》[6],提出“鼓励境外原研药品以国际多中心临床研究的方式,对中国患者人群和境外患者人群同步开展人体PK、PD、PK/PD、有效性和安全性等系统临床试验”。日本国际多中心临床试验的基本流程见图2。
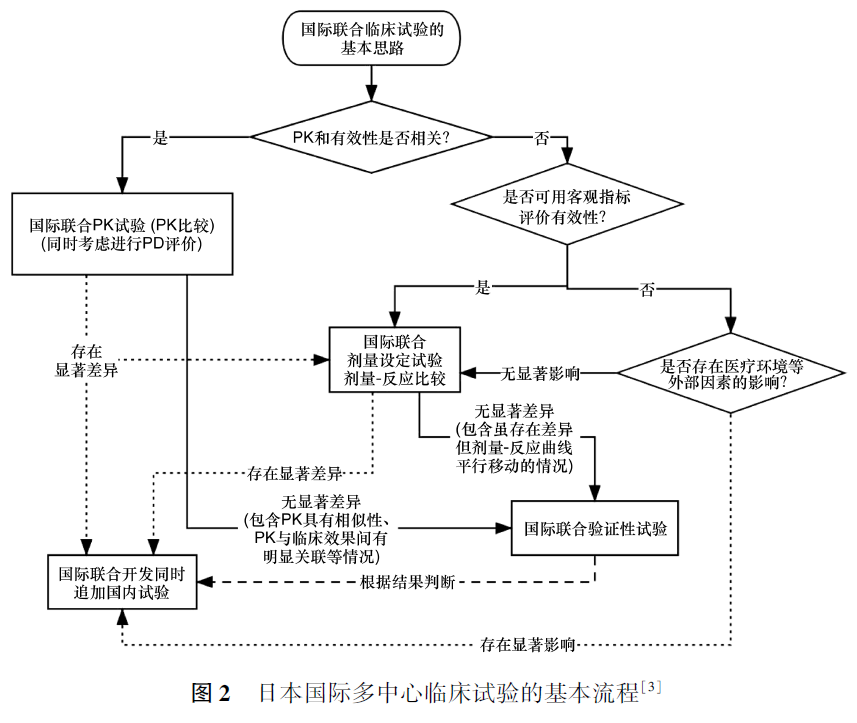
当前,在共同的临床研究方案下进行国际多中心试验,加快完成全球范围内的临床研究,获得尽可能多的地区、不同种类人群的试验数据已成为大势所趋,有助于加快药品上市注册进程。若将国际多中心试验作为桥接研究的数据来源,相比于单一地区的临床试验,在外推试验结果方面能提供更有力的证据。
因此,CDE应鼓励以全球注册为目标的申请人开展国际多中心临床试验。然而,各地区的临床试验并不强制同时开展,可以选取某几个地区开展早期临床试验,以接续的桥接研究证明药物在新地区的有效性以及确定该药物对于种族因素是否敏感。
3.2.1 预先考虑地区差异
进行国际多中心试验有助于评估药物的总体疗效,而如果不同亚组间疗效差异较大,可以考虑排除某些地区或地区内的某个亚组。在试验设计阶段,应预先考虑各地区的差异、如何用内在或外在因素对其进行解释、地区差异对有效性和安全性的潜在影响等。
3.2.2 合理选择受试者
应当采用统一的分类或标准对受试者进行选择———客观指标可采用合规的诊断工具,如生化测试与基因测试、主观指标可在不同地区采用统一的疾病量表。所使用的工具均应经过验证,具有明确的定义和标准,在合格的实验室内实施。
3.2.3 确定验证性试验中的药物剂量
进行PK研究和/或PK⁃PD研究,以确定可能对剂量选择产生影响的地区差异,可以通过单一地区的多种族试验或多地区的单一试验获得PK/PK⁃PD数据。如果存在尚未充分研究的地区差异或种族差异,应当在与试验地区最相关的主要种族群体中进行PK研究。在已知存在药动学差异的亚群之间进行适当的药动学比较,将有助于决定是否需要在不同地区和/或亚群进行药效学研究和剂量反应研究[16]。
3.2.4 科学选择终点指标
主要终点应当与目标人群有关,应当考虑地区内的药物、疾病和人口特征[17]。理想的临床试验终点应当能够检测治疗的预期效果[18],具有临床相关性,被医疗实践所接受(如被监管指南或专业社会指南所接受),并且有足够的敏感性和特异性。
3.3 规范药品上市后研究
对于已经过CDE充分评估,认为符合优先审评审批条件,具有一定疗效且风险基本可控的5.1类新药,可允许申请人进行上市后研究,通常为全国性、多中心、开放标签的研究,用于评估该药物在中国患者中的安全性、疗效、药效学和药动学。然而,应针对具体的评估标准出台相应指南,规范上市后研究的内容和上市后研究的程度等。
4 小结
目前,我国进口药品注册过程中对于境外临床试验数据的接受仍没有严格的标准[19],首先,国家药品监督管理局还应当针对桥接研究出台进一步的指南;其次,缺少豁免进口注册临床试验的技术标准;另外,优先审评药品是否进行上市后研究、上市后研究如何严格监管没有约束性规定。我国还须规范桥接研究的设计,鼓励原研药品通过国际联合试验、多中心试验进行上市注册,逐步规范境外临床试验数据的衔接。
参考文献
详见《中国新药杂志》 2023年第32卷第10期
 产业资讯
珍立拍 2026-05-19
429
产业资讯
珍立拍 2026-05-19
429
 产业资讯
深究科学 2026-05-19
429
产业资讯
深究科学 2026-05-19
429
 产业资讯
中国医药报 2026-05-19
556
产业资讯
中国医药报 2026-05-19
556
 热门资讯
热门资讯 微信公众号
微信公众号