

 产业资讯
产业资讯
 潮药Bar
潮药Bar  2026-05-22
2026-05-22
 405
405
摘要:tarlatamab的获批让DLL3×CD3 TCE站上了SCLC后线的舞台,但随之而来的联用探索——特别是与B7H3 ADC的配合——很快遇到了麻烦:旁观者效应在TCE激活的炎症微环境里被放大成CRS风险,两个分子本该协同,结果互相添乱。
于是有人开始问一个更根本的问题:既然TCE知道怎么找肿瘤,何必非要带着个ADC搭档,为什么不干脆把毒素偶联到TCE分子自己身上?这就是TDC——T cell engager-Drug Conjugate,一个试图同时握住TCE和ADC两把刀的融合型分子。
本文从联用受阻的逻辑开始拆,到TDC安全窗口的工程学本质,最后落在橙帆医药的VBC229和维立志博的LBL-054/LBL-058上——这或许是TCE药物的下一次进化,就像PD-1最后长出了VEGF的另一条臂膀。
小细胞肺癌是肿瘤学里最难攻克的堡垒之一。进展快、转移早、化疗高度敏感却又高度耐药——就像一场节奏过快的火灾,火势汹涌,扑灭容易,但余烬里总藏着复燃的种子。PD-1时代在SCLC这里几乎没留下印记,是DLL3×CD3双抗tarlatamab让这个领域重新看到了光。
2024年tarlatamab在二线及以后SCLC获批,DeLLphi-301研究里ORR接近40%,中位缓解持续时间将近10个月,在这个历史中位生存不足6个月的瘤种里,这组数字已经足够称之为范式性突破了。但医生是贪心的——ORR 40%意味着超过一半的患者没有应答,中位DoR 10个月意味着大多数应答者的缓解终将结束。
于是联用策略自然成了下一步的选项。既然tarlatamab能召唤T细胞,ADC能提供精准的毒素杀伤,两者合流,从理论上讲应该实现TCE重塑免疫微环境、ADC突破抗原异质性的双重叙事。B7H3 ADC当时是最诱人的候选伙伴——B7H3(也称CD276)在SCLC中高度表达,宜联生物的YL201单药数据已经够引人关注,把这两个机制撮合在一起,感觉就差临门一脚了。
结果临床现实给了一个更复杂的答案。tarlatamab联用YL201的探索遭遇阻碍,问题的根源指向B7H3靶点本身的"宽谱性"。B7H3不只在SCLC细胞上高表达,在正常组织中同样有一定程度的分布,而YL201的旁观者效应(bystander killing)在释放毒素时并不加以甄别。当tarlatamab激活T细胞并引发局部细胞因子释放,ADC的bystander效应可能进一步放大免疫激活信号,加剧CRS的风险。
说白了,TCE激活的炎症环境,反而成了ADC毒性放大的温床。两个独立的毒性机制在同一个炎症微环境里叠加,产生的不是协同,是失控的放大效应。这不是联用策略本身有问题,是靶点选错了。B7H3太宽,宽到bystander effect没有边界。爆款联用叙事,被泼冷水
安进和勃林格殷格翰随后的策略调整清楚印证了这个判断——两家公司把视线转向了再鼎医药DLL3 ADC(zoci),尝试以DLL3×CD3 TCE与同靶点ADC联用。逻辑是说得通的:DLL3是SCLC神经内分泌表型的高度特异性靶点,正常组织表达极为有限,旁观者效应有了更明确的边界,毒素更可能被约束在DLL3高表达的肿瘤细胞周围,而不会弥散到正常组织并与TCE激活的免疫浪潮叠加。
但如果根源是"TCE激活的免疫环境放大了ADC的毒性",那联用策略的本质就是把两个独立设计、独立给药、各有毒性窗口的分子强行捆绑在一起,然后期望它们在肿瘤床内精准协同,而不在正常组织中相互叠加。
这是个精密的平衡,但也是个脆弱的不太好把握平衡。两个药的半衰期不同,PK曲线不同,给药节奏不同,靶点竞争不同——要让它们精确地在肿瘤床相遇并协同,本质上是在赌一个很窄的时间窗口(关于两个药联用方面,PK差异的问题是个大问题,这个值得以后单独写篇文章)。
所以有个更根本的问题被问出来了:为什么一定要走联用路线?既然TCE已经知道怎么找到肿瘤细胞,既然ADC的核心价值是把毒素精准送进去——为什么要用两个分子完成一件事,而不是用一个?就像为啥你非要做PD-1联用贝伐珠,而不能造一个AK112成为first in class?
这个问题,就是TDC(T cell engager-Drug Conjugate,T细胞衔接器药物偶联物)这一新型分子设计范式的出发点。TDC的核心设计理念一句话概括:保留TCE的双靶向骨架——一端识别肿瘤抗原(TAA),一端结合T细胞表面的CD3,形成免疫突触激活T细胞——同时在同一骨架上偶联细胞毒性payload,通过TAA端的内吞将毒素递送进肿瘤细胞,发挥ADC式的直接杀伤。
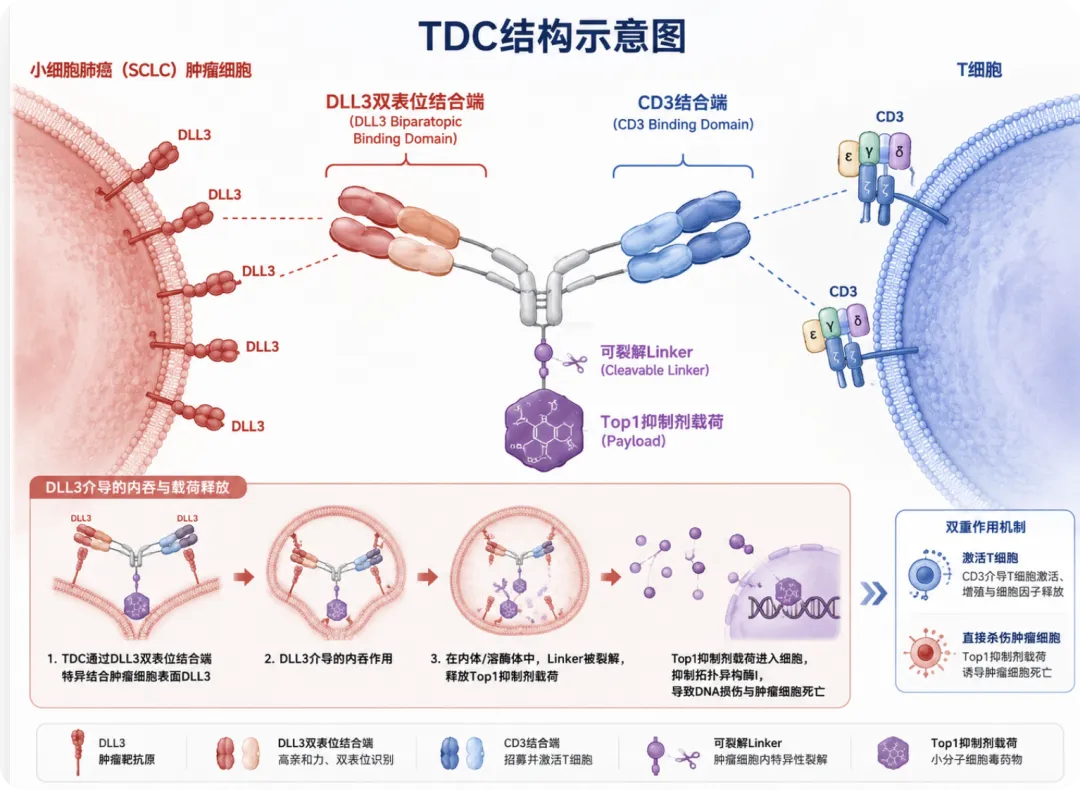
两条杀伤路径天然互补:TCE的免疫杀伤受到抗原异质性限制(DLL3阴性克隆无法被T细胞识别),payload的旁观者效应可以扩大杀伤半径;payload端杀伤遭遇克隆抵抗时,T细胞的持续招募和免疫记忆形成可以延长缓解持续时间。关键是,TDC把这一切整合在单一分子的坐标系内,肿瘤靶向性、免疫激活和毒素递送由同一个分子统一调控,消除了两个独立药物之间PK错位和时间窗口对齐的问题。
但这个设计有一个不可回避的工程学难题。
TDC分子的一端连接CD3,也就是说,它会结合到T细胞表面。这立刻引出了一个让人睡不着的安全性疑问:payload会不会通过CD3端内吞进入T细胞内部,然后毒杀T细胞自身?如果答案是肯定的,TDC的整个逻辑就从根本上坍塌了——它召唤来的T细胞,会被自己携带的毒素消灭。这事儿咋解决,是这个分子模式设计下最根本的问题。
长期以来行业内存在一个直觉性假设:TCR/CD3复合体的内吞效率"天然很低",TDC不需要担心T细胞侧的毒素递送。这个假设是错的。
文献数据说得很清楚:TCR/CD3不是静态膜受体,在静息状态下,TCR:CD3 complex本身就存在速率约1.5%/分钟的基础内吞与回收循环(constitutive internalization and recycling)。
TCR ligation之后CD3表面表达下调的主要机制,也不是内吞速率急剧上升,而是内化的受体被滞留在溶酶体降解路径里出不来(Liu et al., Immunity, 2000)。
Regeneron的研究进一步揭示,CD3 arm亲和力越高,TCE分子越容易进入T细胞的cathepsin B阳性晚期内体/溶酶体区室——亲和力越强,payload释放风险越大;反过来,弱CD3 arm可以显著降低该过程(Haber et al., Scientific Reports, 2021)。
更让人警惕的是体内分布数据——一项CD3×TRP1双抗的研究显示,脾脏(CD3-rich tissue)里的抗体内吞比例约79.7%,比TRP1阳性肿瘤的54.9%还高(Sandker et al., JITC, 2023)。哥们,脾脏内吞比肿瘤还猛,这对TDC来说不是小问题。
说到底,"CD3不内吞"不是一个可以用来托底的假设,而是一个必须被实验数据一条一条驳回的风险点。
所以对TDC而言,更准确的问题框架是什么?我们可以拆解成一套"四级递送漏斗"的逻辑——payload从分子结合到最终在细胞内释放,要经过四道关卡,每道关卡都可以通过分子设计来调节肿瘤细胞侧和T细胞侧之间的比例。
第一级结合(Binding) TDC需要在肿瘤侧实现高亲和力、高avidity结合,同时CD3端保持低亲和力、短驻留时间。CD3 arm的亲和力是细胞因子释放、体内分布和T细胞侧内吞风险的核心调控旋钮,调低它,就是在从源头压低T细胞侧的风险。
第二级内吞(Internalization) DLL3等TAA靶点被结合后往往迅速进入内体路径;通过低亲和力CD3 arm结合的TDC,T细胞侧的内吞量可以通过分子设计压低。Biparatopic设计(双表位锚定同一抗原)能增强avidity并促进受体clustering,进一步加速肿瘤细胞侧的内体化。
第三级释放(Payload Release) 进入细胞不等于payload释放,还要进入晚期内体/溶酶体,linker才会被cathepsin等溶酶体酶裂解激活。低亲和力CD3 arm可以降低TDC在T细胞内到达cathepsin B阳性区室的比例——即便有少量TDC进了T细胞,也可能因为没法进溶酶体而释放不了有效浓度的毒素。
第四级功能性毒性(Functional Toxicity) 最后的安全性验证,必须落到T细胞生存、增殖、功能和细胞因子释放上。T cell viability、γH2AX(DNA损伤标记)、耗竭标志物(PD-1/TIM-3/LAG-3)、PBMC细胞毒性——这些数据才是TDC安全故事的backbone,理论假设只是皮。一个只展示肿瘤杀伤、不展示T cell internalization和PBMC cytotoxicity的TDC项目,安全性故事是不完整的。
TDC安全窗口的核心公式不是"CD3不内吞",而是:TAA端强内吞 × CD3端弱结合短驻留 × linker血浆稳定性 × 溶酶体裂解选择性,共同把肿瘤细胞侧的有效payload递送压倒性地高于T细胞侧。
这套逻辑的第一份临床前实证,来自今天的主角——维立志博的LBL-054和LBL-058。LBL-054是一个CDH17×CD3 TDC,AACR 2026的摘要数据显示,它在肿瘤细胞上表现为strong binding and internalization,在T细胞上却是weak binding、no internalization、no cytotoxicity,同时维持TDCC活性且cytokine release低于传统TCE——这正是"四级漏斗"逻辑的具体落地。
LBL-058则更直接切入DLL3方向,是一个DLL3靶向的T cell engager conjugate,携带Top1 payload,设计上明确采用DLL3高亲和力、CD3低亲和力,目的就是避免T细胞侧binding-mediated cytotoxicity(Sun et al., AACR 2025, Abstract 7322)。两个数据点,把TDC安全窗口从理论推演拉进了可观察的实验框架里。
同一个方向的另一个代表是橙帆医药的VBC229。VBC229是一个DLL3靶向的biparatopic TCE-Top1 inhibitor融合分子,把SCLC领域验证度最高的靶点(DLL3)、最成熟的免疫调动机制(T细胞衔接)和市场验证度最高的payload类别(Topo I抑制剂)整合进同一个骨架——用一个分子,做联用策略试图用两个分子完成的事。
Biparatopic的DLL3设计是关键工程选择:两个不同表位同时锚定DLL3,不只是提高avidity,更重要的是双表位结合能促进受体clustering、加速内吞,为payload递送奠定前提。这一逻辑在DLL3 ADC文献中已有支撑——DB-1314的数据显示DLL3-specific binding可驱动efficient internalization并在SCLC细胞内完成Top1 payload的溶酶体释放。
VBC229目前仍处于临床前,计划2027年上半年提交IND,体内安全窗口、PK特征、CRS风险和T细胞侧payload分布数据还都是待验证项。这个距离临床数据还有相当路程。但它和LBL-054/LBL-058所代表的共同方向——单分子整合TCE和ADC两种能力,而不是靠联用策略在两个独立分子之间寻找协同——在技术路径上有更确定的起点。
结语:一个分子,两把刀。
其实吧回望PD-1的历史,它最大的迭代,也就是从PD-1到PD-1×VEGF,核心是一次分子层面的能力扩展——从单纯解除免疫刹车,到PD-1×VEGF双抗同时解除刹车并切断肿瘤血管新生,一个分子同时执行两个过去需要联用才能完成的功能。这次迭代让PD-1以更强悍的形态延续,在肝细胞癌等适应症里打开了单药联用时代从未打开过的空间。
TCE正在走向类似的路口。tarlatamab的成功证明了DLL3 TCE在SCLC后线的可成药性。但联用ADC的受阻,提示了单纯"召唤T细胞"的边界——DLL3阴性克隆的逃逸、T细胞耗竭,以及在炎症微环境里被放大的CRS风险,都是tarlatamab这一代TCE无法单靠自己解决的问题。
TDC保留了TCE的免疫招募能力,同时赋予它在肿瘤细胞内直接递送毒素的确定性杀伤手段——在一个分子内,同时拥有免疫突触的动态性和payload的确定性。这是联用策略永远无法真正实现的整合。VBC229、LBL-054、LBL-058是否能兑现这个叙事,需要临床数据说话,但它们所代表的方向——让TCE从一把召唤之手,进化为同时能调动免疫系统和直接递送毒素的完整战斗单元——或许正是SCLC后线治疗乃至整个TCE药物开发领域,下一次范式转换的起点。
如果转换成功,那就是从K药到AK112的一次世纪之交般的迭代。
更形象比喻是:从联用到融合,从伴侣到完整体,从召唤者到杀手与召唤者的合体。
上周调研了君实,宜明昂科,以及参加了维立志博的研发日,我对这三个高赔率公司的总结汇报已经放在星球,大家可以观看回放。星球最近做了比较多的大单品财务模型预测,帮助大家更好去预测看biotech的价值。
本周知识星球两场路演:一场是一家美股biotech催化剂与估值模型,周五晚上;一场是必贝特深度拆解,周日晚上。
需要约1V1医药公司具体路演的请进星球,到时候和Jerry预约时间。这周已经被约满啦,得约下周了。所以大家要约的话请尽快来约,要不然得排到下个月啦。
 产业资讯
药智数据 2026-05-22
28
产业资讯
药智数据 2026-05-22
28
 产业资讯
细胞基因治疗前沿 2026-05-22
352
产业资讯
细胞基因治疗前沿 2026-05-22
352
 产业资讯
药智网 2026-05-22
384
产业资讯
药智网 2026-05-22
384
 热门资讯
热门资讯 微信公众号
微信公众号